
A review on the advancement of ether synthesis from organic solvent to water
Sangita Mandal
a,
Swagata Mandal
a,
Sumanta K. Ghosh
a,
Pintu Sar
a,
Aniruddha Ghosh
a,
Rumpa Saha
b and
Bidyut Saha
*a
aHomogeneous Catalysis Laboratory, Department of Chemistry, The University of Burdwan, Golapbag, Burdwan, 713104, WB, India. E-mail: b_saha31@rediffmail.com; Fax: +91-342-2530452; Tel: +91-9476341691 Tel: +91-342-2533913
bDepartment of Chemistry, TDB College Raniganj, Raniganj, West Bengal 713383, India
First published on 21st July 2016
Abstract
Ethers have been synthesized by the different protocols such as Williamson ether synthesis, the Mitsunobu reaction, bimolecular dehydration, the Ullmann method, a transition metal-free coupling reaction between aliphatic alcohols and unsymmetric diaryliodonium salts, room temperature ionic liquid promoted synthesis, Cu(II) catalyzed synthesis, microwave assisted synthesis, and synthesis under solvent free micellar conditions. A good number of homogeneous Bronsted acids and Lewis acid based transition metals have also been reported as catalysts in the etherification of alcohols. The above mentioned pathway has exhibited some drawbacks including their deactivation through decomposition caused by the water formed during the course of the reaction. In many cases these methods also reveal the accumulation of a significant amount of acid at the end of the reaction due to the hydrolysis of Lewis acid catalysts such as metal oxides, which upon neutralization give a considerable amount of salts. A literature survey shows some reports on the use of phase transfer and polymer and clay supported catalysts for the synthesis of symmetrical and unsymmetrical ethers. To a synthetic chemist, an aqueous micellar solution is a good choice as a reaction medium for synthesizing several organic compounds. Micellar conditions are a new direction of study for ether synthesis as they are simple, efficient, economical and environmentally friendly.
 Sangita Mandal | Sangita Mandal was born in Khatra, Bankura in 1991. She passed her M.Sc degree from The University of Burdwan in 2013 and got a UGC-RGNF fellowship in 2015. She also cleared the NET exam in Chemical Sciences in 2012. She is working in a lab in the “Homogeneous Catalysis” division. |
 Swagata Mandal | Swagata Mandal was born in Bankura in 1991. She passed her M.Sc degree from The University of Burdwan in 2013 and got a UGC-RGNF fellowship in 2015. She is working in a lab in the “Homogeneous Catalysis” division. |
 Sumanta K. Ghosh | Sumanta K. Ghosh was born in Burdwan in 1974. He passed his M.Sc degree from Visva-Bharati in 2010 and got a NET-CSIR fellowship in 2001. He was awarded a Ph.D. degree from the University of Burdwan in 2013. He is now working as a Post Doc. |
 Pintu Sar | Pintu Sar was born in Hooghly, in 1989. He passed his M.Sc degree from Visva-Bharati in 2011 and got a NET-UGC fellowship in 2012. He is presently working in the lab. He is working in the “Homogeneous Catalysis” division. |
 Aniruddha Ghosh | Aniruddha Ghosh was born in Raniganj, in 1988. He passed his M.Sc degree from The University of Burdwan in 2010 and got a NET-UGC fellowship in 2010. He is working as a SRF in the lab. He is working in the “Homogeneous Catalysis” division. |
 Rumpa Saha | Rumpa Saha was born in Burdwan, 1987. She passed her M.Sc. degree from the University of Burdwan, 2009 and got a CSIR NET fellowship in 2009. She was awarded a Ph.D. degree from the University of Burdwan in 2015. Dr Rumpa Saha is presently working as an Assistant Professor in the Department of Chemistry, TDB College, Raniganj, Burdwan, India. |
 Bidyut Saha | Bidyut Saha was born in Birbhum, WB, India in 1975. He obtained his Ph.D degree from Visva Bharati University, India in 2007. He was a visiting scientist for the year 2009–2010 in the Department of Chemistry, UBC, Vancouver, Canada. Dr Saha is presently working as an Associate Professor in the Department of Chemistry, The University of Burdwan, India. His area of interests is bioremediation of toxic metals, micellar catalysis and chemical kinetics. He has already published 90 papers in international journals. |
1. Introduction
Ethers are a class of organic compounds characterized by an oxygen atom bonded to two alkyl or aryl groups (e.g. R–O–R′). Generally ethers may be categorized as aryl ether, allyl ether, phenolic ether, phenacyl ether, and polyether. They have different structures depending upon the groups that are connected to the ether oxygen. Crown ethers are special type of cyclic ether. These types of ethers are low molecular polyethers.1 The compounds containing an ether oxygen with a phenyl group on one end and aryl, alkyl, benzyl, allyl, vinyl, etc. on the other end are called phenolic ethers or aromatic ethers. These ethers have a wide range of applications. The uses of phenolic ethers depend upon the nature of the group attached to the other end of the phenolic oxygen. For example, diphenyl ether and its halo derivatives have agrochemical applications such as herbicides and fungicides.2,3 Similarly phenolic ethers find applications in chemical engineering,4 pharmaceuticals, and food.5The preparation of ethers is an important synthetic reaction for which a wide variety of procedures have been developed during the last hundred years. The Williamson reaction, discovered in 1850, is still the best general method for the preparation of unsymmetrical as well as symmetrical ethers.6–10 Williamson ether synthesis is an organic reaction, forming ether from an organic halide and an alcohol. The discovery, development and identification of biologically active compounds has gained a lot of importance in recent years. Even though there are a considerable number of adverse effects, chemists have always tried to design a drug possessing maximum therapeutic application and minimum toxicity.11 Organic ethers are one of the most important classes of chemicals which have significant applications as herbicides, disinfectants, pharmaceuticals, plasticizers, solvents, drug intermediates and as solvents in organic synthesis. They are also used as important precursors for polymers and fragrances.12,13
A good number of homogeneous Bronsted acids and Lewis acid catalysts are commonly used in the preparation of unsymmetrical substituted ethers.14 There are few available procedures for the conversion of phenols or alcohols into ethers: Mitsunobu-type rections,15 reductive condensation of ketones and esters,16 hydroalkonylation of alkynes,17 and oxidative C–H alkoxylation of arenes.18 The methods above can be considered as a general method for the preparation of ethers.
Another general root for the synthesis of aryl ethers is typically achieved via a nucleophilic aromatic substitution (NAS) process or transition metal catalyzed Ullmann type reaction.19,20 Remarkable drawbacks encountered in many conventional methods necessitated the search for an appropriate reaction system to enhance reaction conversion and lessen environmental pollution. Combinatorial synthesis has brought about a lot of changes in the recent trends of ether synthesis in aqueous micellar media. Many chemical reactions with organic substances conducted in the laboratory as well as in industry need organic solvents as reaction media, even though water is safe, benign, environmentally friendly and cheap compared with organic solvents such as DMF, DMSO, DCM, etc. In spite of all these factors, the goal is to develop a novel catalytic system that uses water as a solvent for a wide range of reactions. A perusal of the literature indicated that surfactants can be used to promote a variety of synthetic organic reactions. It is believed that surfactants form micelles in protic solvents such as water which act as micro reactors to enhance the reaction rates and afford very good to excellent yields of end products. Micelles behave as nano-reactors for metal based oxidation reactions such as Cr(VI),21–24 Ce(IV),25–28 Mn(VII),29 V(V).30–32
Dimethyl ether (DME) is the simplest ether, which can be synthesized from methanol dehydration over an acid support or from syngas conversion via a two step reaction with methanol as an intermediate. DME can also used as LPG, diesel fuel, etc.33
2. Solvents for ether synthesis
Many chemical reactions with organic substances conducted in the laboratory as well as in industry need organic solvents as reaction media. In ether synthesis the solvent which can be used is water34 because water is safe, benign, environmentally friendly and cheap compared with organic solvents35 and another two reasons are:(i) Most of the organic substances are insoluble in water and soluble in a polar organic solvent.
(ii) A lot of organic substrates and catalysts are decomposed or deactivated in aqueous media.
The dehydration of alcohol is a fundamentally important organic transformation that is being widely used for the synthesis of ether compounds in both industrial and academic laboratory settings.
3. Physical properties of ethers
At room temperature most ethers are a colorless, sweet-smelling liquid. The C–O bonds in ethers are polar and thus ethers have a net dipole moment. The weak polarity of ethers does not appreciably affect their boiling points which are comparable to those of the alkenes of comparable molecular mass. Ethers have much lower boiling points as compared to isomeric alcohols. This is because alcohol molecules are associated by hydrogen bonds while ether molecules are not.364. Several methods for synthesis of ethers
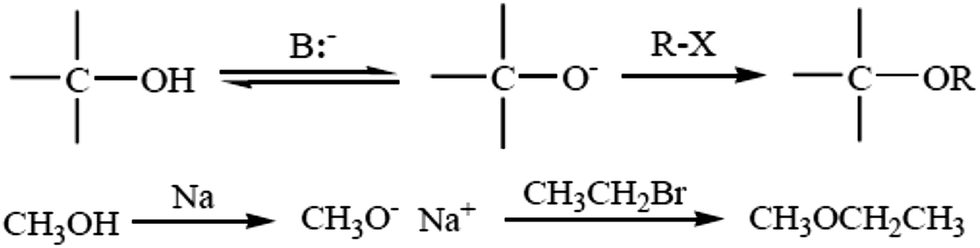
4.1. Williamson ether synthesis
The most versatile method for making ethers is Williamson ether synthesis (Scheme 1), named after the English chemist Alexander Williamson, who devised the method in the 19th century.37 The Williamson reaction is a conventional method for transformation in organic synthesis since the products are of value in both academic and industrial applications. The Williamson synthesis usually involves the employment of an alkali-metal salt of the hydroxy compound and an alkyl halide. These reactions are usually performed using organic solvents in the presence of an inorganic base followed by refluxing for several hours.38 There are a few useful procedures available for the conversion of phenols into aromatic ethers, which do not require the initial formation of a phenoxide ion.39 Phenols are not only important building blocks for constructing pharmaceuticals, polymers, and natural compounds, but also serve as versatile synthetic intermediates in preparing oxygenated heterocycles.40 This reaction is important in the history of organic chemistry because it helped to prove the structure of ethers. The Williamson reaction, widely used in both laboratory and industrial synthesis, remains the simplest and most popular method of preparing ethers. Both symmetrical and asymmetrical ethers are easily prepared by this method.41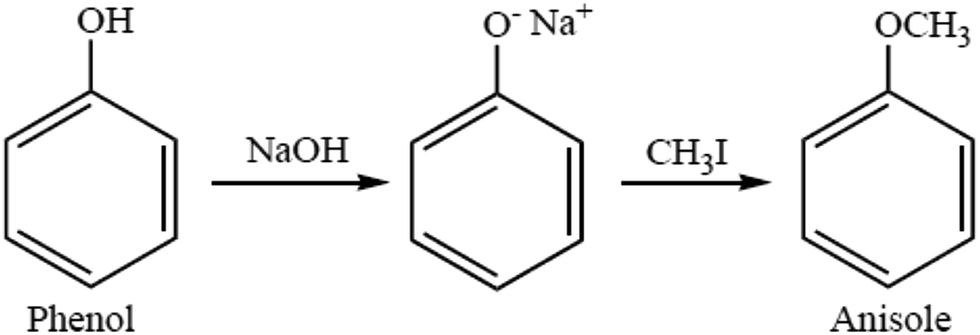 | ||
| Scheme 1 Williamson ether synthesis. | ||
There are several examples given below for the synthesis of ethers:
4.2. Bimolecular dehydration
In the presence of acid two molecules of an alcohol may lose water to form an ether. In practice, however, this bimolecular dehydration to form ether competes with unimolecular dehydration to give an alkene (Scheme 2). Bimolecular dehydration produces useful yields of ethers only with simple, primary alkyl groups such as those in dimethyl ether and diethyl ether.42 Dehydration is used commercially to produce diethyl ether. This process is very much useful because the byproduct is water.43 For a dehydration reaction in which water molecules are generated during the reaction, the water molecules must be removed to shift the equilibrium to the dehydrated product side.44Examples given in Scheme 2.
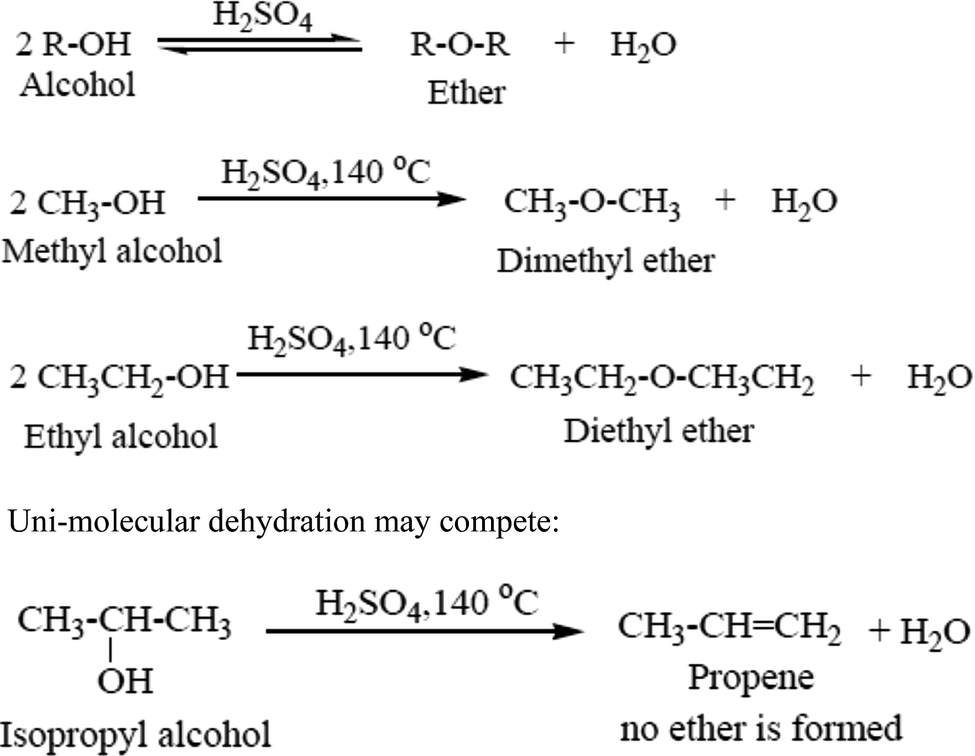 | ||
| Scheme 2 Bimolecular dehydration ether synthesis. | ||
4.3. Ullmann ether synthesis
Low molecular weight polyphenyl ether is usually prepared using the Ullmann ether synthesis. The synthesis of aryl ethers is typically achieved via a nucleophilic aromatic substitution (NAS). The NAS approach requires the use of phenoxide to displace aryl fluorides or chlorides that are activated by strong electron withdrawing groups, EWG, located in the ortho, para, or, in rare cases, meta positions.45 Due to the high costs associated with palladium based catalyst systems, in the past decade the development of a multitude of copper catalysts for Ullmann-type O-arylation46–55 reactions has been thoroughly reviewed.20 The effectiveness of these catalyst systems for the synthesis of aryl ethers is highly dependent on the substrate, ligand, solvent, base, and reaction temperature. During the course of a recent investigation of poly(arylene ether) synthesis, it became necessary to prepare a variety of asymmetric diaryl ether monomers and we chose to take advantage of the versatility of O-arylation reactions.56–62 Copper catalyzed O-arylation reactions (Scheme 3) are typically carried out in polar, aprotic solvents. However, with the requirement of utilizing non-polar solvents, the use of toluene and xylene was explored. There are limited reports of Cu-catalyzed O-arylation reactions being performed in toluene or xylene. To that end we screened the catalytic behavior of a wide range of air stable, readily available and low cost Cu(I) complexes: CuXPPh3 and CuX(PPh3)3 where X = Cl, Br, or I.63 A variety of reaction conditions were studied in order to evaluate the effects of the catalyst,64 base, solvent, reaction temperature and electronic properties of both the substrate and phenoxide ion.65 An example of Ullman ether synthesis is shown in Scheme 3.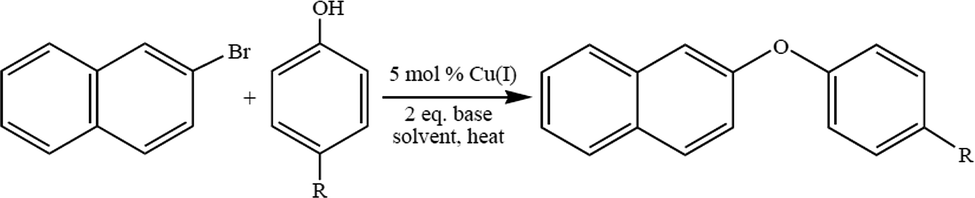 | ||
| Scheme 3 Ullmann ether synthesis. | ||
4.4. Transition metal-free synthesis of alkyl–aryl ethers
The synthesis of alkyl–aryl ethers provides access to industrially important compounds in a sustainable manner. A wide range of alkyl–aryl ethers have been produced by using easily assembled unsymmetric diaryliodonium salts (Scheme 4) in combination with primary, secondary, tertiary, allylic, and benzylic aliphatic alcohols. The utilization of an inexpensive, broadly useable and recoverable auxiliary renders this strategy sustainable and practically attractive.66 The formal synthesis of pioglitazone further demonstrates the synthetic utility of this method.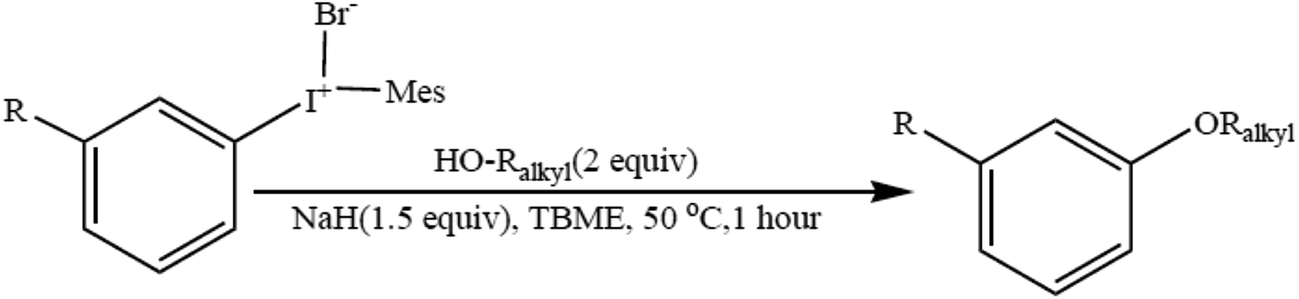 | ||
| Scheme 4 Transition metal-free coupling ether synthesis. | ||
4.5. Room-temperature ionic liquid-promoted Williamson synthesis of ethers
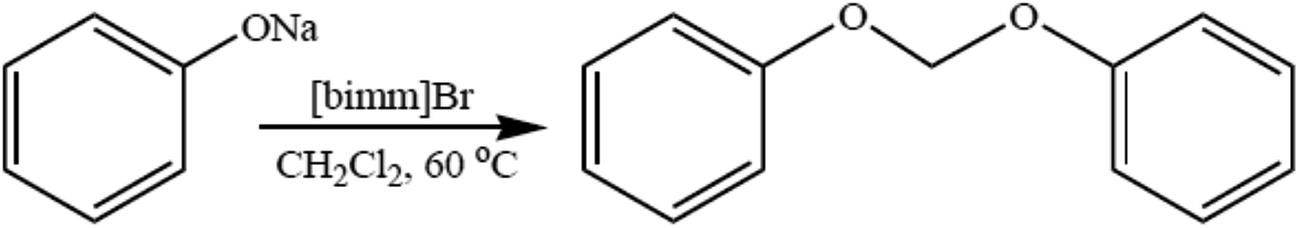
The development of environmentally friendly catalysts and solvents for organic chemistry is an area of considerable importance. Diaryloxymethanes are synthetically important compounds used as fungicides,60 parasiticides,61 in the treatment of liver fluke infection in mammals,62 and in the preparation of condensation polymers.63,64 Diaryloxymethanes were used in the preparation of various catalysts such as cavitations65 and crown ethers66 and in the preparation of diols such as 2,2-o-dihydroxy-6,6-o-dimethoxy-1,10-biphenyl. Diaryloxymethanes were prepared through methylation of the appropriate phenol with diiodomethane, bromochloromethane, and dichloromethane using different inorganic bases, such as Cs2CO3, KOH, and NaH.63–67 The reactions of alkali metal phenolates with methylating agents were generally carried out in several polar solvents such as DMSO and DMF. An ionic liquid was used for regioselective O-alkylation of naphthols, although C-alkylation also occurs with this method which requires a prolonged time period and harsh reaction conditions.65 Recently room-temperature ionic liquids as “green solvents” in organic synthesis have gained considerable importance because of their solvating ability, negligible vapor pressure, and easy recyclability and reusability.68These unique properties of ionic liquids make them a strong alternative to conventional solvents. Herein we used the ionic liquid 1-methyl-3-butylimidazolium bromide [bmim]Br for the reaction of phenoxides with alkyldihalides. The ionic liquid [bmim]Br was prepared as per the literature method.69–72 Substituted phenoxide ions react with dichloromethane in the presence of the ionic liquid [bmim]Br at 60 °C without any added catalyst (Scheme 5). The synthesized diaryloxymethanes were fully characterized by IR, 1H-NMR, and 13C-NMR, and physical constants were assigned with their reported values in the literature. The ionic liquid was recovered (almost 100%) and reused at least three times (Table 1) for the reaction of p-methoxyphenoxide with dichloromethane without any considerable loss in yield.67
 | ||
| Scheme 5 Room-temperature ionic liquid-promoted ether synthesis. | ||
| Run | Yield (%) |
|---|---|
| 1 | 90 |
| 2 | 89 |
| 3 | 88 |
| 4 | 8 |
5. Catalysis in ether synthesis
5.1. Copper(II)-catalyzed ether synthesis from aliphatic alcohols and potassium organotrifluoroborate salts
The formation of carbon–heteroatom bonds using metal catalysis is emerging as one of the most significant classes of cross-coupling reactions. In the area of C–O ether bond formation, there has been a resurgence of interest in Cu mediated reactions beyond that of the classical Ullmann ether synthesis.73,74 A protocol for the Cu(II) catalyzed etherification of aliphatic alcohols under mild and essentially neutral conditions has been described. Air and moisture stable potassium alkenyl and aryl trifluoroborate salts undergo cross-coupling with a variety of aliphatic primary and secondary alcohols and phenols, and are tolerant of a range of functional groups. The optimized conditions utilize catalytic copper(II)acetate with 4-dimethylamino pyridine as a ligand in the presence of 4 Å molecular sieves under an atmosphere of oxygen (Scheme 6).75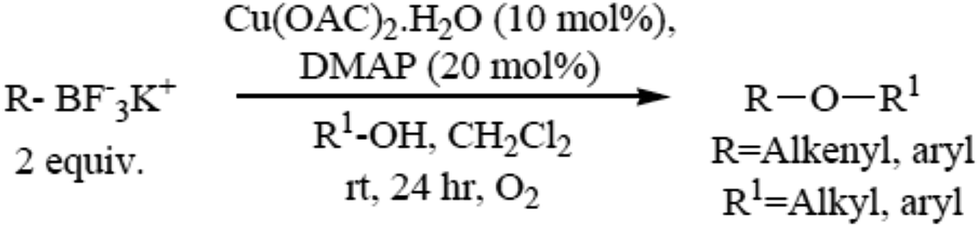 | ||
| Scheme 6 Copper catalyzed ether synthesis. | ||
5.2. Microwave-assisted synthesis of diaryl ethers without a catalyst
Diaryl ether motifs are known to be present in a variety of natural products and biologically interesting compounds.76 We have also reported that microwave-assisted coupling of phenols, including those having a strong electron withdrawing group, with aryl halides in the presence of potassium carbonate provide diverse diaryl ethers within a few minutes.77 A range of phenols were employed to couple with the electron deficient aryl halides (Scheme 7) such as mono-halogen substituted benzonitrile and 1-chloro-4-nitrobenzene in the presence of 2 equiv. of potassium carbonate under microwave irradiation in a DMSO medium and the results were obtained.78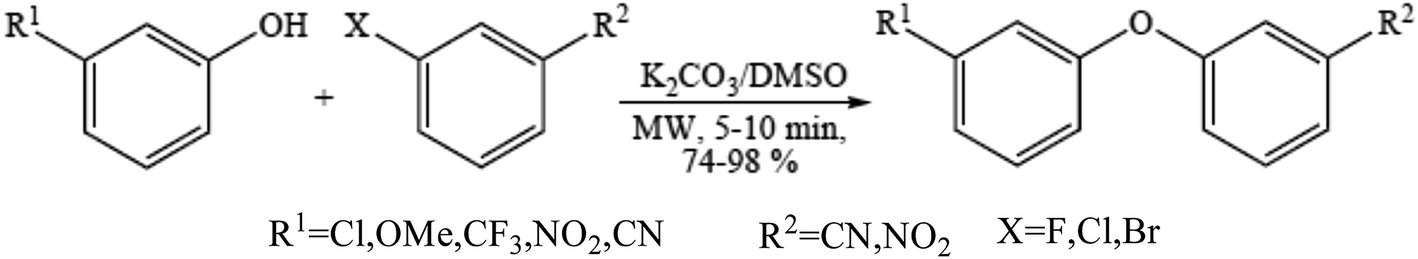 | ||
| Scheme 7 Microwave-assisted synthesis without catalyst. | ||
5.3. Ether synthesis under solvent-free micellar condition
A good number of homogeneous Bronsted acids and Lewis acid based transition metals have also been reported as catalysts in the etherification of alcohols. However, these systems exhibited some drawbacks including their deactivation through decomposition by the water formed during the course of reaction. In many cases these methods also reveal the accumulation of a significant amount of acid at the end of the reaction due to the hydrolysis of Lewis acid catalysts such as metal oxides, which upon neutralization give a considerable amount of salts. The acid waste, residual catalyst and salt material may cause several environmental problems when they are disposed off, and some of them may be toxic. Difficulties such as catalyst recovery for reuse and neutralization of the reaction mixture also make the process tedious. A literature survey shows some reports on the use of phase transfer, and polymer and clay supported catalysts for the synthesis of symmetrical and unsymmetrical ethers.79–85 Another general root for the synthesis of organic ethers is the Mitsunobu reaction in which PPh3 or polymer bound PPh3 is used as the catalyst.15 Triphenylphosphene is highly sensitive86,87 to water and toxic in nature and the reaction83 demands inert and dry conditions.88,89A literature survey indicated that surfactants have been used to promote a variety of synthetic organic reactions. It is believed that surfactants form micelles in protic solvents such as water which act as micro reactors90,91 to enhance the reaction rates and afford very good to excellent yields of end products.92–94 One of the main areas for achieving this target is the exploration of alternative reaction conditions and reaction media to accomplish the desired chemical transformations with minimized byproducts or waste as well as eliminating the use of conventional organic solvents, wherever possible. Consequently, several newer strategies have appeared such as solvent free reactions (grinding), multi-component reactions under solvent free conditions could enhance the efficiency from an economic as well as an ecological point of view and so the solid state organic reactions are gaining significance both from mechanistic and synthetic points of view.95,96 Synthesis of naphthol ethers under Williamson conditions requires several hours (≥20 h) at relatively high temperature. Although there are some reports to modify the drastic conditions of these reactions, many of them exhibited long reaction times and accumulation of unwanted byproducts, which ultimately involved tedious work-up procedures. Encouraged by this aspect, we conducted an O-alkylation of aromatic and heteroaromatic alcohol97 reaction in a micellar medium under acid-free conditions.11
The literature also indicated11 that etherification of phenols and hydroxy pyridine can be performed using two methods; one is acidic conditions and the other is replacing H2SO4 with a variety of micelle forming surfactants (acid-free conditions) such as sodium dodecylsulphate (SDS), cetyltrimethylammonium bromide (CTAB), and Triton X-100 (TX-100). When β-naphthol reacts with methanol in acidic conditions, the time required is 20 hours, yield 91%, but in acid free conditions when CTAB is used the time required is only 1.1 hours, yield 96%, in the case of SDS the time required is 2.0 hours, yield 86% and for TX-100 the time required is 3.0 hours, yield 91%. The results indicated that the catalytic activity of different micelles in etherification is in the order: CTAB > SDS > TX-100 (Scheme 8).
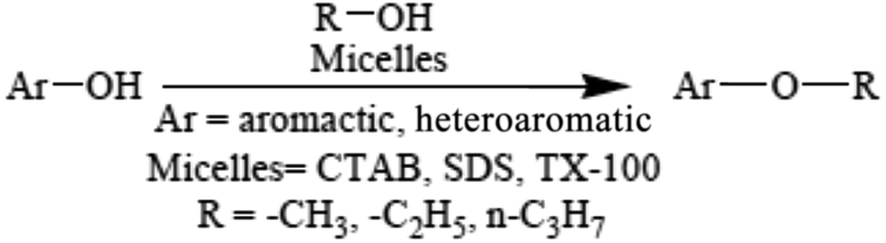 | ||
| Scheme 8 Etherification in aqueous micellar media. | ||
The catalytic effect of the micellar solution of CTAB may be attributed to the hydrophobic nature of organic substrates (Scheme 9).
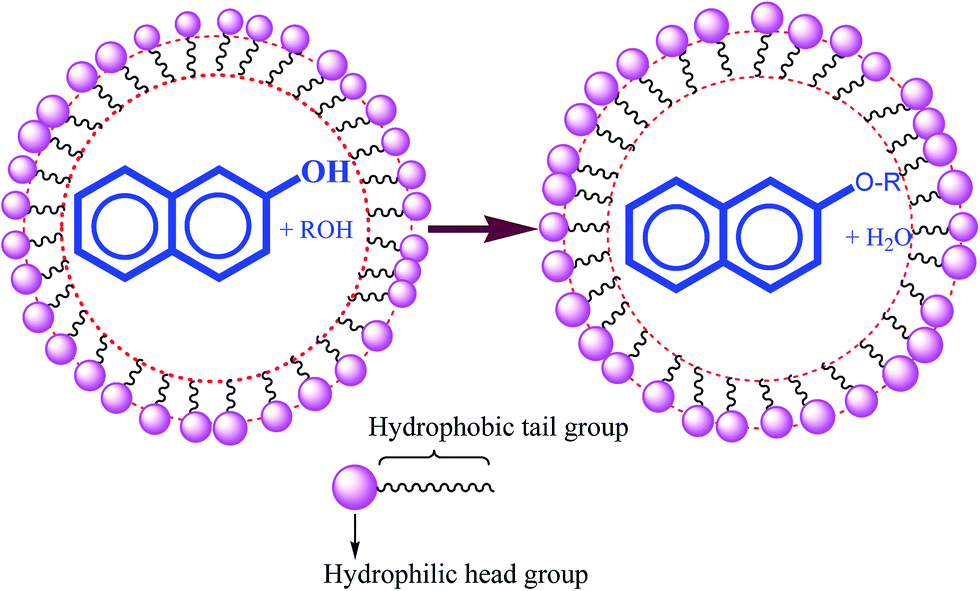 | ||
| Scheme 9 Localization of reactant molecules in micellar media. | ||
6. Comparison of experimental details for etherification of β-naphthols
6.1. Experimental procedure for etherification of β-naphthols under conventional conditions
Substituted β-naphthol (1.44 g, 10 mmol) was added to a stirred solution of methanol (0.64 g, 20 mmol) and 0.1 M micellar solution (10 mL) and then heated up to reflux. The progress of the reaction was monitored by TLC. After the completion of the reaction, the reaction mixture was diluted with dichloromethane (10 mL), and separated from the aqueous layer. The organic layer was then washed with water (3 × 5 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford the crude product. The crude product was purified by column chromatography (silica gel, 100–200 mesh) using EtOAc–hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) as an eluent to obtain the desired product.11
:9) as an eluent to obtain the desired product.11
6.2. Experimental procedure for etherification of β-naphthols by ultrasonically assisted etherification (USAS)
Substituted β-naphthol (1.44 g, 10 mmol) was added to a solution of methanol (0.64 g, 20 mmol) and 0.1 M micellar solution (10 mL) at room temperature, which was then sonicated at 40 °C using an ultrasonic bath. The ultrasonic bath had a frequency of 33 kHz and an electric power rating of 100 W. The reaction was carried out in a round bottom flask of 50 mL capacity equipped with a mechanical agitator and the flask was suspended at the centre of the ultrasonic bath. The final products were isolated by absorbing the reaction mixture into silica gel and purifying it by column chromatography using an ethyl acetate/hexane gradient.116.3. Experimental procedure for etherification of β-naphthols by microwave irradiated etherification (MWAR)
Substituted β-naphthol (1.44 g, 10 mmol) was added to a solution of methanol (0.64 g, 20 mmol) and 0.1 M micellar solution (10 mL) and these mixtures were heated using a controlled microwave synthesizer (Biotage Initiator + SP Wave model, 0–200 W at 2.45 GHz, capped at 60 W during steady state) for a few minutes (attains a temperature of 120 °C and 1 bar pressure). The final products were isolated by absorbing the reaction mixture into silica gel and purifying it by column chromatography using an ethyl acetate/hexane gradient.117. Disadvantages of conventional method and advantages of aqueous method
The disadvantages of this method are the very dilute solutions (3 to 12% strength aqueous solutions), the sometimes long reaction times of up to 24 hours and the associated low yield. The low selectivities are likewise disadvantageous with the platinum catalysts used;98 the yields are only ca. 68 to 89% following workup by distillation. The Williamson reaction often competes with the base catalyzed elimination of the alkylating agent, and the nature of the leaving group as well as the reaction conditions (particularly the temperature and solvent) can have a strong effect on which is favored. In particular, some structures of alkylating agent can be particularly prone to elimination. When the nucleophile is an aroxide ion, the Williamson reaction can also compete with alkylation on the ring since aroxide is an ambident nucleophile.99The reaction times were drastically reduced to one to two hours under sonication and few minutes under microwave conditions from the several hour timescales of classical reactions. Thus, the present protocols show rate acceleration associated with high product yields, when compared with similar reactions performed under classical conditions.11 Water is not only an inexpensive and environmentally benign solvent, but also plays an important role in reactivity and selectivity. Surfactants catalyze the reaction efficiently with short reaction times without using any harmful organic reagents and solvents.
8. Industrial advances and future prospects
8.1. Solvent
Ethers are quite unreactive and therefore desirable solvents for fats, oils, waxes, perfumes, resins, dyes, gums, and hydrocarbons. In addition, vapours of certain ethers are used as insecticides and fumigants for top soil.1008.2. Medicinal purposes
Ethers also play an important role in pharmacology and medicine, especially for the use of anesthetics101 (Fig. 1a). Methoxymethane otherwise known as codeine is an effective pain-relieving drug, and is the primary ingredient in morphine. Ethyl ether (or just ether) was one of the first anesthetics replaced by other less-flammable anesthetics, such as nitrous oxide and halothane used for surgical procedures during the 1800s. A mixture of alcohol and ether was known as "Spirit of ether" or Hoffman's drops though it is flammable. | ||
| Fig. 1 Several uses of ether. | ||
8.3. Industrial purposes
Ethoxy ethane (ethyl ether) is quite volatile and therefore used as a starter fluid for diesel engines (Fig. 1b) and gasoline engines in cold weather. Dimethyl ether is used as a spray propellant and refrigerant; ethers of ethylene glycol are used as solvents and plasticizers. Methyl t-butyl ether (MTBE) is a gasoline stabilizer which increases octane concentration and reduces pollutants.1028.4. Dimethyl ether use, production, and application
Most of the above fundamental studies were performed on a laboratory scale, whereas outstanding development for DME synthesis has also been completed on an industrial scale in recent years. Regarded as a clean fuel103 for the 21st century, DME has remarkable market and industrial potential, for electric power generation and as a home energy source,104 for tasks such as heating and cooking, and is greatly cost-competitive with LPG and diesel fuel (Fig. 1c).37 Ethers are also important in medicine and pharmacology, especially for use as anesthetics.9. World consumption of ethyl ether in 2013
The United States is the leading producer of ethyl ether accounting for about 88% of its production in all of North and South America in 2013. The production of ethyl ether in Mexico, Argentina and Brazil is minor, while Canada imports its ethyl ether from the United States.105The following pie diagram (Chart 1) shows the world consumption of ethyl ether:
 | ||
| Chart 1 World consumption of ethyl ether. | ||
The main applications in the United States continue to be for use as engine starting fluids and for smokeless gunpowder, which together account for more than two-thirds of consumption. However, limited growth is expected in these areas. Relatively stronger growth will occur for laboratory and industrial reagent use, which currently accounts for 25% of domestic ethyl ether use. Overall, consumption of ethyl ether in North and South America is expected to grow slightly106 by 2% annually during 2013–2018.
10. Conclusion
The present review should demonstrate that surfactant associates considerably extend organic chemistry in water and lead to notable effects on the rate of reaction. This write up aims to bring the attention of scientists working in the field of industrial chemistry, green chemistry, and catalysis towards the potential future of ether synthesis in micellar media. Aqueous surfactant media catalyze the reaction efficiently with short reaction times without using any harmful organic solvents and afford very good to excellent yields of end products. Surfactants also help to solubilize organic substrates in water. Apart from discovering new or improving existing organic transformations, it will surely give us a glimpse into the fascinating and still poorly understood world of water. Like a growing child, organic synthesis in water still surprises us by most unexpected questions and observations. In reality despite the original results micellar catalysis is only occasionally employed above laboratory scale. The limited applicability to organic chemistry has its origin in the problematic work-up of the reactions, since separation of the aqueous phase and organic phase is difficult.References
- G. K. D. Devi and M. I. F. Mohamed, Chem. Sin., 2012, 3, 71–75 Search PubMed.
- A. Prasad, K. S. Nimavat and K. B. Vyas, Chem. Sin., 2011, 2, 26 CAS.
- M. Nallu, R. Selvakumar and M. Krishnapillai, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1999, 38, 71–75 Search PubMed.
- A. A. Moroz and M. S. S. Russian, Chem. Rev., 1974, 43, 679–689 Search PubMed.
- R. Kahn, I. Low and H. Trishman, Chem. Ber., 1957, 90, 296–300 CrossRef.
- Comprehensive Organic Chemistry, ed. N. Baggett, D. Barton, W. D. Ollis and J. F. Stoddart, Pergaman, Oxford, 1st edn, 1979, p. 819 Search PubMed.
- J. March, Advanced Organic Chemistry, Wiley, New York, 4th edn, 1992, p. 386 Search PubMed.
- S. Kim, K. N. Chung and S. Yang, J. Org. Chem., 1987, 52, 3917–3919 CrossRef CAS.
- K. J. Miller and M. M. AbuOmar, Eur. J. Org. Chem., 2003, 7, 1294–1299 CrossRef.
- C. Siswanto, T. Battal, O. E. Schuss and J. F. Rathman, Langmuir, 2013, 23, 6047–6052 Search PubMed.
- K. R. Reddy, K. C. Rajanna, S. Ramgopal, M. S. Kumar and S. Sana, Green Sustainable Chem., 2012, 2, 123–132 CrossRef.
- W. H. Miles and K. B. Connell, J. Chem. Educ., 2006, 83, 285–286 CrossRef CAS.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. D. Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS PubMed.
- N. Baggett, in Comprehensive Organic Chemistry, ed. D. Barton, W. D. Ollis and J. F. Stoddart, Pergamon, Oxford, 1st edn, 1979, p. 819 Search PubMed.
- K. C. K. Swamy, N. N. B. Kumar, E. Balaraman and K. V. P. P. Kumar, Chem. Rev., 2009, 109, 2551–2651 CrossRef CAS PubMed.
- N. Sakai, T. Moriya and T. Konakahara, J. Org. Chem., 2007, 72, 5920–5922 CrossRef CAS PubMed.
- M. C. Haibach, C. Guan, D. Y. Wang, B. Li, N. Lease, A. M. Steffens, K. K. Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2013, 135, 15062–15070 CrossRef CAS PubMed.
- X. Wang, Y. Lu, H. X. Dai and J. Q. Yu, J. Am. Chem. Soc., 2010, 132, 12203–12205 CrossRef CAS PubMed.
- R. Frlan and D. Kikelj, Synthesis, 2006, 14, 2271–2285 Search PubMed.
- J. S. Sawyer, Tetrahedron Lett., 2000, 41, 5045–5065 CrossRef.
- R. Saha, A. Ghosh and B. Saha, Chem. Eng. Sci., 2013, 99, 23–27 CrossRef CAS.
- S. K. Ghosh, R. Saha, K. Mukherjee, A. Ghosh, S. S. Bhattacharyya and B. Saha, J. Korean Chem. Soc., 2012, 56, 164 CrossRef CAS.
- K. Mukherjee, R. Saha, A. Ghosh, S. K. Ghosh and B. Saha, J. Mol. Liq., 2013, 179, 1–6 CrossRef CAS.
- P. Sar, A. Ghosh, S. Malik and B. Saha, J. Carbohydr. Chem., 2016, 35, 86–105 CrossRef CAS.
- A. Ghosh, P. Sar, S. Malik and B. Saha, J. Mol. Liq., 2015, 211, 48–62 CrossRef CAS.
- S. Malik, A. Ghosh, K. Mukherjee and B. Saha, Tenside, Surfactants, Deterg., 2014, 51, 325–332 CrossRef CAS.
- A. Ghosh, R. Saha and B. Saha, J. Mol. Liq., 2014, 196, 223–237 CrossRef CAS.
- A. Ghosh, R. Saha, K. Mukherjee, P. Sar, S. K. Ghosh, S. Malik, S. S. Bhattacharyya and B. Saha, J. Mol. Liq., 2014, 190, 81–93 CrossRef CAS.
- A. Ghosh, K. Sengupta, R. Saha and B. Saha, J. Mol. Liq., 2014, 198, 369–380 CrossRef CAS.
- B. Saha, Inorg. React. Mech., 2008, 6, 287–291 CAS.
- B. Saha, S. Sarkar and K. M. Chowdhury, Int. J. Chem. Kinet., 2008, 40, 282–286 CrossRef CAS.
- B. Saha, K. M. Chowdhury and J. Mandal, J. Solution Chem., 2008, 37, 1321–1328 CrossRef CAS.
- J. Sun, G. Yang, Y. Yoneyama and N. Tsubaki, ACS Catal., 2014, 4, 3346–3356 CrossRef CAS.
- A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725–748 CrossRef CAS PubMed.
- B. H. Lipshutz and S. Ghoraib, Green Chem., 2014, 16, 3660–3679 RSC.
- A. W. Bosman, H. M. Janssen and E. W. Meijer, Chem. Rev., 1999, 99, 1665–1688 CrossRef CAS PubMed.
- A. Williamson and L. Justus, Ann. Chem., 1851, 77, 37–49 CrossRef CAS.
- E. Fuhrmann and J. Talbiersky, Org. Process Res. Dev., 2005, 9, 206–211 CrossRef CAS.
- P. S. Fier and J. F. Hartwig, Angew. Chem., Int. Ed., 2013, 52, 1002–1009 CrossRef PubMed.
- J. H. P. Tyman, Synthetic and Natural Phenols, Elsevier, New York, 1st edn, 1996, p. 598 Search PubMed.
- L. Krti and B. Czako, Strategic Applications of Named Reactions in Organic Synthesis, Elsevier, Burlington, 2005, pp. 484–485 Search PubMed.
- W. Yang, J. Xia, B. Zhang, Q. Song, Q. Chen and D. Mao, J. Catal., 2005, 230, 140–149 CrossRef.
- F. Cunill, M. Iborra, J. F. Izquierdo, C. Fité and J. Tejero, J. Mol. Catal. Chem., 2002, 182, 541–554 Search PubMed.
- K. Manabe, S. Iimura, M. S. Xiang and S. Kobayashi, J. Am. Chem. Soc., 2002, 124, 11971–11978 CrossRef CAS PubMed.
- Z. Yu, L. S. Tan and E. Fossuma, ARKIVOC, 2009,(xiv), 255–265 CAS.
- J. W. W. Chang, S. Chee, S. Mak, P. Buranaprasertsuk, W. Chavasiri and P. W. Chan, Tetrahedron Lett., 2008, 49, 2018–2022 CrossRef CAS.
- W. Chen, J. Li, D. Fang, C. Feng and C. Zhang, Org. Lett., 2008, 10, 4565–4568 CrossRef CAS PubMed.
- R. Gujadhur and D. Venkataraman, Synth. Commun., 2001, 31, 2865–2879 CrossRef CAS.
- R. K. Gujadhur, C. G. Bates and D. Venkataraman, Org. Lett., 2001, 3, 4315–4317 CrossRef CAS PubMed.
- R. Hosseinzadeh, M. Tajbakhsh, M. Mohadjerani and M. Alikarami, Synlett, 2005, 7, 1101–1104 CrossRef.
- D. Ma and Q. Cai, Org. Lett., 2003, 5, 3799–3802 CrossRef CAS PubMed.
- A. B. Naidu, O. R. Raghunath, D. J. C. Prasad and G. Sekar, Tetrahedron Lett., 2008, 49, 1057–1061 CrossRef CAS.
- J. Niu, H. Zhou, Z. Li, J. Xu and S. Hu, J. Org. Chem., 2008, 73, 7814–7817 CrossRef CAS PubMed.
- L. W. Xu, C. G. Xia, J. W. Li and X. X. Hu, Synlett, 2003, 20(8), 59–871 Search PubMed.
- T. Schareina, A. Zapf, A. Cotte, N. Muller and M. Beller, Tetrahedron Lett., 2008, 49, 1851–1855 CrossRef CAS.
- F. H. Jardine, L. Rule and A. G. Vohra, J. Chem. Soc. A, 1970, 238–240 RSC.
- A. L. Casado and P. Espinet, Organometallics, 2003, 22, 1305–1309 CrossRef CAS.
- P. F. Barron, J. F. Dyason and P. C. Healy, J. Chem. Soc., Dalton Trans., 1987, 35, 1099–1106 RSC.
- S. K. Sundalam and D. R. Stuart, J. Org. Chem., 2015, 80, 6456–6466 CrossRef CAS PubMed.
- A. Arnoldi, R. Cavzaniga, G. Morini, L. Merlini and G. J. Favina, J. Agric. Food Chem., 2000, 48, 2547–2555 CrossRef CAS PubMed.
- L. R. Jeppson, J. Econ. Entomol., 1946, 39, 813–822 CrossRef CAS PubMed.
- M. Harfenist, Ger. Offen., 1972, 2, 570 (Chem. Abstr., 1972, 77, 19404) Search PubMed.
- C. Creber, Makromol. Chem., 1956, 17, 154–155 Search PubMed.
- M. Minoro, Y. Tsugashi, K. Teruo, M. Shigeru and O. Sabaro, J. Polym. Sci., Part C: Polym. Symp., 1968, 6, 683 Search PubMed.
- R. Esteban, P. Carlos, M. Sandra and E. K. Angel, J. Org. Chem., 1999, 64, 2577–2578 CrossRef.
- H. Henning and U. Rolf, J. Org. Chem., 1992, 57, 5809–5810 CrossRef.
- S. V. More, S. S. Ardhapure, N. H. Naik, S. R. Bhusare, W. N. Jadhav and R. P. Pawar, Synth. Commun., 2005, 35, 3113–3118 CrossRef CAS.
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772–3789 CrossRef CAS.
- T. Walton, Chem. Rev., 1999, 99, 2071–2084 CrossRef.
- M. J. Earle and K. R. Seddon, Pure. Appl. Chem., 2000, 72, 1391–1398 CrossRef CAS.
- G. M. Gordan, Appl. Catal., 2001, 222, 101–117 CrossRef.
- R. Sheldon, Chem. Commun., 2001, 182, 2399–2407 RSC.
- F. Theil, Angew. Chem., Int. Ed., 1999, 38, 2345–2347 CrossRef CAS.
- J. Hassan, M. Sevignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1469 CrossRef CAS PubMed.
- T. D. Quach and R. A. Batey, Org. Lett., 2003, 5, 1381–1384 CrossRef CAS PubMed.
- K. C. Nicolaou and N. C. B. Christopher, J. Am. Chem. Soc., 2002, 122, 5560–5562 Search PubMed.
- F. Li, Q. Wang, Z. Ding and F. Tao, Org. Lett., 2003, 5, 169–2171 CrossRef PubMed.
- G. Mann and J. F. Hartwig, Tetrahedron Lett., 1997, 38, 8005–8008 CrossRef CAS.
- T. A. Rao, D. Dutta and G. I. Georg, Tetrahedron Lett., 1998, 39, 8751–8754 CrossRef.
- A. Corma and M. Renz, Angew. Chem., Int. Ed., 2007, 46, 298–300 CrossRef CAS PubMed.
- K. T. V. Rao, P. S. N. Rao, P. S. Saiprasad and N. L. Gaiah, Catal. Commun., 2009, 10, 1394–1397 CrossRef.
- J. Pozniczek, A. Micekllnicka, A. Lubanska and A. Bielanski, Appl. Catal., A, 2005, 286, 52–60 CrossRef CAS.
- T. Ollevier and T. M. Mwene-Mbeja, Tetrahedron Lett., 2006, 47, 4051–4055 CrossRef CAS.
- Y. Q. Cao and B. G. Pei, Synth. Commun., 2000, 30, 1759–1766 CrossRef CAS.
- T. Mitsudome, T. Matsuno, S. Sueoka, T. Mizugaki, K. Jitsukawa and K. Kaneda, Green Chem., 2012, 14, 610–613 RSC.
- H. Nie, J. Geng, J. Jing, Y. Li, W. Yang and X. Zhang, RSC Adv., 2015, 5, 97121–97126 RSC.
- K. C. Kumara Swamy, N. N. Bhuvan Kumar, E. Balaraman and K. V. P. P. Kumar, Chem. Rev., 2009, 109, 2551–2651 CrossRef PubMed.
- M. Debabrata and S. L. Buchwald, J. Org. Chem., 2010, 75, 1791–1794 CrossRef PubMed.
- M. D. Maiti, Chem. Commun., 2011, 47, 8340–8342 RSC.
- M. H. Mondal, A. Roy, S. Malik, A. Ghosh and B. Saha, Res. Chem. Intermed., 2015, 112, 92707–92718 Search PubMed.
- S. De, S. Malik, A. Ghosh, R. Saha and B. Saha, RSC Adv., 2015, 5, 65757–65765 RSC.
- J. H. Fendler and E. H. Fendler, Catalysis in micellar and macromolecular systems, Academic, New York, 1975 Search PubMed.
- D. A. Sabatini, R. C. Knox and J. H. Harwell, ACS Symp. Ser., 1995, 594, 265–278 CrossRef.
- F. M. Menger, J. U. Rhee and H. K. Rhee, J. Org. Chem., 1975, 40, 3803–3805 CrossRef CAS.
- N. B. Singh, R. J. Singh and N. P. Singh, Tetrahedron Lett., 1994, 50, 6441–6493 CrossRef CAS.
- G. R. Desiraju, Solid State Ionics, 1997, 101–103, 839–842 CrossRef CAS.
- S. K. Sundalam and D. R. Stuart, Org. Process Res. Dev., 2015, 19, 1924–1935 CrossRef.
- V. K. Ahluwalia and R. Aggarwal, Organic Synthesis: Special Techniques, CRC Press, Delhi, 2001 Search PubMed.
- K. Vollhardt, C. Peter and N. E. Schore, Organic Chemistry: Structure and Function, W. H. Freeman and Company, New York, 2007 Search PubMed.
- R. L. Smith, Environ. Health Perspect., 1984, 57, 1–4 CrossRef CAS PubMed.
- J. W. Hill and D. K. Kolb, Chemistry for changing times, Pearson Prentice Hall, Upper saddle river, New Jersey, 10th edn, 2004, p. 257 Search PubMed.
- L. Wang, S. Yu, Z. Mi and H. fang, React. Kinet. Catal. Lett., 2002, 77, 147 CrossRef CAS.
- T. A. Semelsberger, R. L. Borup and H. L. Greene, J. Power Sources, 2006, 156, 497–511 CrossRef CAS.
- V. V. Galvita, G. L. Semin, V. D. Belyaev, T. M. Yurieva and V. A. Sobyanin, Appl. Catal., A, 2001, 216, 85–90 CrossRef CAS.
- Chemical Economics Handbook, Glycol Ethers, Published March 2014 Search PubMed.
- J. Mark, J. L. Guardia, C. Robert and B. Hale, J. Am. Chem. Soc., 2013, 17, 9643–9650 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |