
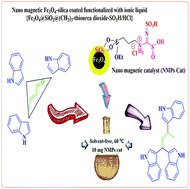
Synthesis of the first magnetic nanoparticles with a thiourea dioxide-based sulfonic acid tag: application in the one-pot synthesis of 1,1,3-tri(1H-indol-3-yl) alkanes under mild and green conditions†
Abstract
Novel and recyclable thiourea dioxide-based magnetic nanoparticles with a sulfonic acid tag {Fe3O4@SiO2@(CH2)3–thiourea dioxide–SO3H/HCl} are described. The described catalyst was fully characterized using Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD) patterns, scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDX), transmission electron microscopy (TEM), thermogravimetric analysis (TGA), atomic force microscopy (AFM) and ultraviolet-visible spectroscopy (UV/Vis). The reported novel magnetic nanocatalyst presents an excellent activity and catalytic performance for the synthesis of 1,1,3-tri(1H-indol-3-yl)alkane derivatives through one-pot three-component mixed Mannich-type and Friedel–Crafts reactions under solvent-free conditions at 60 °C.
 Please wait while we load your content...
Please wait while we load your content...

