
A facile one step route to synthesize WO3 nanoplatelets for CO oxidation and photodegradation of RhB: microstructural, optical and electrical studies†
Abstract
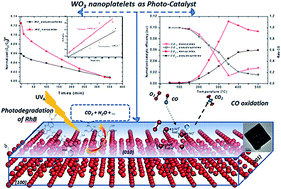
Two-dimensional nanoplatelets of tungsten trioxide (NP-WO3) were synthesized at room temperature, using a wet chemical method, without any surfactants or templates; aqueous mineralization was obtained by simply adjusting the pH. The resulting nanostructures were characterized using X-ray diffraction combined with Rietveld refinements, Raman and UV-Vis spectroscopies. Their morphologies and sizes were analyzed by scanning and electron microscopies. The electrical, optical, catalytic and photocatalytic properties of the NP-WO3 nanoplatelets were then determined and compared to the ones of pseudospherical (PS-WO3) standard samples. Nanoplatelets as well as pseudospherical particles crystallized in the single orthorhombic WO3 phase. The Rietveld refinement calculations and HRTEM analyses revealed a strong distortion of the WO6 octahedra, according to the W–O splitting. The electrical conductivity of WO3 compact pellets showed that both samples were semi-conducting with a weak difference in activation energies. Using a homemade photocatalytic device, the NP-WO3 particles used as photocatalyst in an aqueous medium, exhibited a significant efficiency to decompose rhodamine B over their large exposed surface (010), compared to PS-WO3 particles. These NP-WO3 particles were also used as the catalytic material for oxidation in an air–CO gas flow. They exhibited catalytic activity higher than the one in the PS form.
 Please wait while we load your content...
Please wait while we load your content...

