
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural evolution induced preferential occupancy of designated cation sites by Eu2+ in M5(Si3O9)2 (M = Sr, Ba, Y, Mn) phosphors†
Yi.
Wei‡
a,
Chun Che
Lin‡
*c,
Zewei
Quan
d,
Maxim S.
Molokeev
 ef,
Victor V.
Atuchin
ghi,
Ting-Shan
Chan
j,
Yujun
Liang
a,
Jun
Lin
*b and
Guogang
Li
*a
ef,
Victor V.
Atuchin
ghi,
Ting-Shan
Chan
j,
Yujun
Liang
a,
Jun
Lin
*b and
Guogang
Li
*a
aFaculty of Materials Science and Chemistry, China University of Geosciences, Wuhan 430074, P. R. China. E-mail: ggli8312@gmail.com
bState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: jlin@ciac.ac.cn
cCondensed Matter and Interfaces, Debye Institute for Nanomaterials Science, Utrecht University, Princetonplein 5, 3584 CC Utrecht, The Netherlands. E-mail: cclin0530@gmail.com
dDepartment of Chemistry, South University of Science and Technology of China, Shenzhen, Guangdong 518055, P. R. China
eLaboratory of Crystal Physics, Kirensky Institute of Physics, Siberian Branch of the Russian Academy of Sciences, Krasnoyarsk 660036, Russia
fDepartment of Physics, Far Eastern State Transport University, Khabarovsk 680021, Russia
gLaboratory of Optical Materials and Structures, Institute of Semiconductor Physics, SB RAS, Novosibirsk 630090, Russia
hFunctional Electronics Laboratory, Tomsk State University, Tomsk 634050, Russia
iLaboratory of Semiconductor and Dielectric Materials, Novosibirsk State University, 2 Pirogov Str., Novosibirsk 630090, Russia
jNational Synchrotron Radiation Research Center, Hsinchu 300, Taiwan
First published on 10th June 2016
Abstract
In this paper, we present new insight into a changing Eu2+ crystallographic site preference in Eu-doped M5(Si3O9)2 (M = Sr, Ba, Y, Mn), which is related to the structural variation induced by M cation substitutions. The effect of the local structural geometry on the luminescence properties of Eu2+ is revealed. By substitution of Ba2+ for Sr2+, the lattice expansion is restricted to specific cation sites, resulting in the abrupt blue shifted emission of Eu2+ ions. The abnormal blue shift on replacing Sr2+ with Mn2+ is attributed to the preferential 6-fold coordination for Mn2+ that moves the Eu2+ ions to other sites. The results elucidate the mechanisms of emission band adjustment by local site coordination change and it can be potentially extended to crystals which properties are sensitive to local lattice variations.
Rare-earth-activated silicates are widely used as phosphor materials for white light-emitting diode (WLED) illumination because of their high quantum efficiency and low cost.1–3 However, unsatisfactory thermal stability and spectral position of the excitation/emission bands limit the applications of these materials. To overcome these disadvantages, different cation substitutions were used to adjust the spectroscopic parameters of silicate phosphors, and numerous corresponding mechanisms were proposed to elucidate the relationship of the structural and luminescence properties. These mechanisms include the “cation size mismatch”, “neighboring cation substitution”, “nanosegregation and neighbor cation control” and “chemical unit co-substitution” effects.4–12 However, these mechanisms cannot ultimately solve all problems, and many questions remain unanswered. To date, the cation substitution effect, which can systematically tune spectral position and thermal quenching by changing the coordination environment of the selected cation sites and controlling the preferential activator ion occupancies, has rarely been considered. In the present study, we present a new insight into the structural variation induced site-preferential occupancy of Eu2+ in M5(Si3O9)2 (M = Sr, Ba, Y, Mn) phosphors through size-mismatched cation substitutions applicable to spectral positions and thermal stability tuning.
In this study, the Sr2.97−xBaxEu0.03Y2(Si3O9)2 (Ba series) and Sr2.97−yMnyEu0.03Y2(Si3O9)2 (Mn series) solid solutions (0 ≤ x ≤ 1.59, 0 ≤ y ≤ 0.63) were successfully prepared. The doping concentration of Eu2+ was controlled at the level of 1 at% of Sr2+ in the Sr3Y2(Si3O9)3 host. The synthesis route and characterization description can be found in ESI.† Firstly, the phase composition and structural properties of the samples were identified by XRD analysis. For Ba series, all diffraction peaks of the compounds with x = 0, 0.06 and 0.09 were indexed in the monoclinic cell (C2/c) of Sr3Y2(Si3O9)3, as plotted in Fig. S1 (ESI†) and Fig. 1a.13 A similar C2/c cell was obtained for the composition range of 0.18 < x < 1.59. However, a noticeable difference was observed in the diffraction patterns in comparison with those recorded from the low-doped samples (Fig. S1 (ESI†)), implying a possible phase transition induced by the substitution of Ba2+ for Sr2+. Accordingly, these phases can be reasonably differentiated as Phase 1 (0 ≤ x ≤ 0.09) and Phase 2 (0.18 < x ≤ 1.59). Samples with x = 0.12, 0.15, 0.18 represent the mixture of these two phases. Notably, the pure phase state of the Ba series is destroyed at x > 1.59, and many impurities appeared, including, besides (Sr,Ba)3Y2(Si3O9)2, the known silicates BaSi2O5, Ba5Si8O21 and BaY2Si3O10 (Fig. S1 (ESI†)). Thus, the structural analysis was mainly focused on the evolution from Phase 1 to Phase 2.
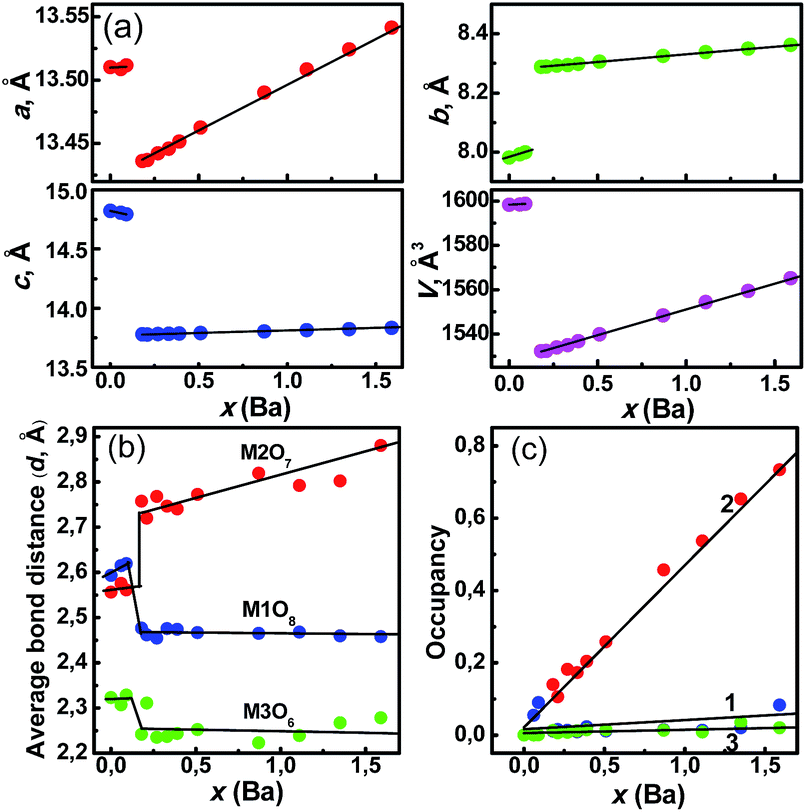 | ||
Fig. 1 (a) Dependence of cell parameters a, b, c and cell volume V on concentration x (Ba) in Sr3−xBaxY2(Si3O9)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Eu (0 < x ≤ 1.59); dependence on concentration x (Ba) of: (b) average bond length: d(M1–O) (M1O8), d(M2–O) (M2O7), d(M3–O) (M3O6); (c) occupancies of M1 site (line 1), M2 site (line 2) and M3 site (line 3) by Ba ions. :Eu (0 < x ≤ 1.59); dependence on concentration x (Ba) of: (b) average bond length: d(M1–O) (M1O8), d(M2–O) (M2O7), d(M3–O) (M3O6); (c) occupancies of M1 site (line 1), M2 site (line 2) and M3 site (line 3) by Ba ions. | ||
The evident chemical shift from ∼604.2 to ∼650.4 cm−1 at x ∼ 0.15 was observed for the representative band from the Raman spectra of the Ba series, verifying the phase transition emergence, as shown in Fig. S2 (ESI†). To determine the effect of this phase transition on the coordination environment of M (Sr/Ba/Y/Eu) ions, Rietveld refinement was performed for the Ba series. The crystal structure of Sr3Y2(Si3O9)2 was taken as a starting point,13 The refinement parameters and cell parameters a, b, c, and V are presented in Table S1, Fig. S3–S17 (ESI†), and Fig. 1a. The chemical composition of the compounds was close to the nominal compositions, as obtained by the inductively coupled plasma element analysis (Table S2, (ESI†)). Thus, the total site occupancies were constrained during refinement in accordance to the designed compositions. As shown in Fig. 1a, the linear increase of cell parameters and volume on the Ba concentration increase at x < 0.09 and x > 0.18 in accordance with Vegard's rule proves the solid solution formation at Phase 1 and 2 ranges. The cell parameter jump appeared at x ∼ 0.15. The a and c values markedly decreased at x ∼ 0.15, whereas b increases abruptly.
Unexpectedly, V noticeably decreases at x ∼ 0.15, as the Ba concentration increase should imply a cell volume increase. This situation is unusual and it may lead to an unexpected change in the coordination environment of Eu2+.
According to the refinement results (Table S1, (ESI†)), three cation sites M1, M2 and M3 (M1:M2:M3 = 2:2:1) in M5(Si3O9)2 are coordinated with eight (M1O8), seven (M2O7) and six (M3O6) oxygen atoms, respectively. The calculated average bond lengths (d) for the M1, M2 and M3 sites are shown in Fig. 1b. At x ≤ 0.09, all bond lengths slightly increase when the smaller Sr2+ ions are gradually substituted with larger Ba2+ ions. This result is attributed to the random distribution of Ba2+ ions over the three sites in Phase 1. Further, at 0.09 < x ≤ 0.18, d(M2–O) exhibits a sharp increase from 2.56 Å to 2.75 Å, while d(M1–O) (2.62 → 2.48 Å) and d(M3–O) (2.32 → 2.24 Å) decrease. Consequently, the polyhedra M1O8 and M3O6 decrease in size, and only M2O7 polyhedron expands during phase transition. In Phase 2, only d(M2–O) increases with x, whereas both d(M1–O) and d(M3–O) basically remain constant. This result indicates that Ba2+ ions preferentially substitute Sr2+ ions at M2 sites. The opposite bond length changes during phase transition are responsible for the cell volume decrease (Fig. 1a). In addition, the dependence of occupancies in the three sites was investigated. It is clearly shown in Fig. 1c (line 2) that only M2 sites were occupied by Ba2+ ions after the phase transition, whereas M1 and M3 were preferential sites for Y3+ ions. To emphasize the main features of phase transition associated with three M (Sr/Ba/Y/Eu) sites, the model of structural transformation was built, as shown in Fig. 2a and b. Sr3Y2(Si3O9)2:Eu consists of Sr/Y/Eu atom layers and Si3O9 ring layers.14 The cube, monocapped trigonal prism and octahedron for M–O share two O atoms with each other. Owing to the phase transition, M1O8 changes from the cube to square antiprism because of the several M1–O bond rotations. This finding indicates a more compact Si3O9 unit packing around the M1 sites with reducing d(M1–O) and the volume (Fig. 1b). The preferential occupation of M3O6 octahedron by smaller Y3+ ions results in a shrinkage of this polyhedron.
 | ||
| Fig. 2 Mechanism of crystal structure transformation from (a) Sr3Y2 (Si3O9)2:Eu (Phase 1, x = 0) to (b) Sr3−xBaxY2(Si3O9)2:Eu (Phase 2, x = 1.59). | ||
Monocapped trigonal prism M2O7 is markedly shrank along the c-axis direction from 2.67 Å to 2.42 Å (Fig. 1b). All these shrinkages in the M sites finally lead to the cell volume decrease and a shrinkage between two SiO4 polyhedra. Although the shrinkage occurs along the c-axis of the M2O7 polyhedron, the final d(M2–O) increases and is, particularly, increasing abruptly during phase transition. This abnormal phenomenon originates from the expansion along the a- and b-axes. This situation also causes the expansion between two SiO4 polyhedra along the a-axis direction. Generally, with the Ba2+ content increase from the phase transition point, only M2O7 polyhedra expand, whereas M1O8 and M3O8 polyhedra shrink regardless of a final increase in the cell volume. Therefore, Ba2+ ions preferably occupy the M2 site, whereas the M1 and M3 sites are preferential for Y3+ ions.
The 5d–4f transition of Eu2+ is sensitive to the structural variation of the host lattice that even a slight change in the local coordination environment around Eu2+ can cause a big effect for its luminescence.15–20 The photoluminescence excitation (PLE) and emission (PL) spectra of the Ba series are shown in Fig. 3a and b, respectively. For x = 0, the PLE consists of a broad band from 250 to 430 nm with the maximum at 365 nm. Under 365 nm UV, a bluish-green emission is given with CIE color coordinates (0.168, 0.258). The PL spectrum covers a broad range from 425 to 575 nm, centered at 474 nm. The asymmetric emission results from the three Sr2+ sites available for Eu2+, as shown by the three Gaussian fitting peaks at 21978 cm−1 (455 nm), 20833 cm−1 (480 nm), and 19231 cm−1 (520 nm) at M1, M2 and M3 sites (Fig. 3b). With the Ba2+-doping in Phase 1, a slight blue shift emission from 474 nm (x = 0) to 469 nm (x = 0.09) was observed, as shown in Table S3 and Fig. S18 (ESI†). This shift is attributed to random occupation of larger Ba2+ over Sr2+ sites in Phase 1 and the resulting cell enlargement. Thus, the crystal field splitting (CFS) of Eu2+ 5d energy levels in these enlarged sites weakened, resulting in blue shift emission. During the phase transition, the large blue shift emission from 468 to 438 nm occurred. The result implied that the lattice environment around Eu2+ became looser, and thus the average Eu–O bond length (d) increased. Generally, the crystal field strength is proportional to 1/d5 Therefore, Eu2+ ions in the looser sites with the longer bond length will possess a higher energy emission and it will generate a blue shift of the emission band.
 | ||
| Fig. 3 (a) The photoluminescence excitation (PLE) and (b) photoluminescence emission (PL) spectra of Sr2.97−xBaxEu0.03Y2(Si3O9)2 (x = 0–1.59) recorded at maximal emission and excitation wavelengths, respectively. The emission peak of Sr2.97Eu0.03Y2(Si3O9)2 was decomposed by Gaussian fitting. (c) The dependence of the emission peaks of Ba series on concentration x (Ba). | ||
In view of the size-difference between Ba2+ ions and Sr2+ ions, the coordination environment of M ions would change with the Ba2+ doping, which could be depicted by the change of d(M–O). The d(M–O) values at different Ba-doping concentration can be calculated on the base of XRD measurements of the studied samples. According to the refinement results, d(M2–O) increases from 2.56 Å to 2.75 Å on the Ba content increase in the transition phase, whereas d(M1–O) and d(M3–O) slightly decrease. Therefore, Eu2+ ions should occupy the looser M2 sites and generate a large decrease in CFS of 5d energy levels of Eu2+, leading to the abnormal large-scale blue shift. This evident blue shift emission during phase transition can be more clearly observed from the color coordinates: (0.161, 0.205) for x = 0.09 and (0.157, 0.073) for x = 0.21. Given 0.18 < x ≤ 1.59, the crystal structure of the Ba series stabilizes at Phase 2, and the small blue shift from 438 nm at x = 0.18 to 432 nm at x = 1.59 mainly originates from a continuous decrease in CFS of the 5d energy levels of Eu2+ in view of the gradual increase of d(M2–O) with x. Given x > 1.59, the stable structure was destroyed, and numerous foreign phases appeared. Hence, certain low-energy emission peaks emerged, generating a broadened emission and red shift, as shown in Fig. S19 (ESI†). Generally, a large spectral blue shift in the Ba series was induced by the change in the crystal field environment at specific cation sites because of the phase transition. Thus, it offers a novel and efficient route to tune the optical properties of phosphor materials.
On the basis of the same principle, a spectral red shift can be expected in the Mn series because of the enlarged CFS of Eu2+ by the substitution of smaller Mn2+ for larger Sr2+. Actually, unusual blue shifts in the Mn series spectra were observed with the Mn2+ content (y) increase, as shown in Fig. 4a and b. To reveal the mechanism of the luminescence blue shift, the phase purity and structure variation in the Mn series were first analyzed by XRD and Rietveld refinement. Evidently, the Mn series samples crystallize in the monoclinic Sr3Y2(Si3O9)2 phase, space group C2/c, and the XRD diffraction peaks continuously shift to larger angles with the y increase, as shown in Fig. S20 (ESI†). This is in agreement with Vegard rule, revealing the solid solution formation.4 For the Mn series, the refinement results are presented in Table S4 and Fig. S21–S26 (ESI†). The results verify the generation of host-type structures due to Mn/Eu doping. The cell volume decrease with the y increase reflects the formation of Mn series solid solutions (Fig. S27 (ESI†)).
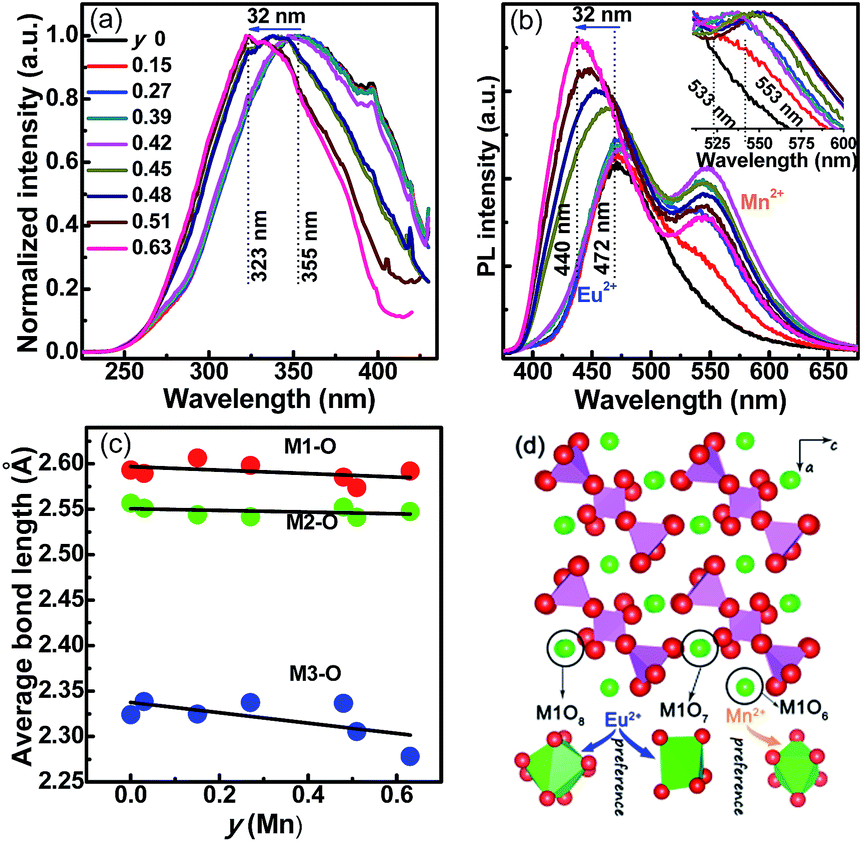 | ||
| Fig. 4 (a) PLE and (b) PL spectra of Sr2.97−yMnyEu0.03Y2(Si3O9)2 (y = 0–0.63, Mn series) samples. The normalized PL spectra ranging from 510 nm to 600 nm of Mn2+ emission are shown in the insert. (c) Average bond length (Å) [d(M1–O), d(M2–O) and d(M3–O)] as a function of doping concentration y (Mn). (d) A schematic explanation of the preferential occupancy of Eu2+ and Mn2+ ions in Mn series. | ||
As shown in Fig. 4a, at y < 0.45, the PLE spectra of the Mn series are similar to those of the Ba series, except for the maximum at 355 nm. The PL spectra consist of two broad bands in the range of 400–650 nm, which are centered at 472 nm and 545 nm, respectively. The first band is attributed to the 5d–4f transition of Eu2+, whereas the second band is related to the 4T1g(4G)–6A1g(6S) transition of Mn2+. Notably, the emission positions of the Eu2+ and Mn2+ bands remain persistent up to Mn2+ doping content y ∼ 0.45. However, the emission intensity of the Mn2+ band gradually increased with the y increase, whereas the Eu2+ emission intensity first increases and then decreases. Thus, a tunable single-composition white light emission can be obtained by mixing blue-green light (Eu2+) and yellow light (Mn2+). Beyond y = 0.45, sudden and converse emission shifts for Eu2+ (blue shift) and Mn2+ (red shift) were observed, as shown in Fig. 4b. The red shift of Mn2+ emission is reasonable, as the cell volume of Mn series shrinks with the y increase because of the smaller Mn2+ size in comparison with Sr2+, thus enlarging the CFS of the Mn2+ 5d levels. However, evident blue shifts of Eu2+ emission appeared in the PLE and PL spectra, shifting to 32 nm (355 → 323 nm) and 32 nm (472 → 440 nm), respectively. The results indicate that Mn2+ and Eu2+ ions exhibit preferential lattice site occupancies in the Mn series solutions. The average M–O bond lengths as a function of y in the Mn series are arranged in Fig. 4c. With changing y from 0 to 0.63, only d(M3–O) exhibits the shrinkage, whereas both d(M1–O) (a slight decrease) and d(M2–O) are almost constant. This implies that Mn2+ ions are more easily coordinated with six oxygen atoms, following the priority of MnO6 > MnO8 > MnO7. Therefore, a possible mechanism is proposed for the abnormal blue shift of Eu2+ emission (Fig. 4d). At low Mn2+ doping levels (y < 0.45), Eu2+ and Mn2+ ions randomly enter M1–M3 sites. At y > 0.45, the d(M3–O) is lower than 2.33 Å, which is smaller than the sum of Eu2+ and O2− ion radii [r(Eu2+) + [6]r(O2−) = 1.17 Å + 1.4 Å = 2.57 Å], but larger than the sum of Mn2+ and O2− ion radii [r(Mn2+) + [6]r(O2−) = 0.67 Å + 1.4 Å = 2.07 Å][6]. Therefore, Mn2+ ions preferentially occupy the M3 sites, driving Eu2+ ions to the M2 and M1 sites. As mentioned in the previous section, the Eu–O bond length significantly affects the crystal field strength (Dq), that is, Dq is proportional to 1/d5, and the looser site accommodating Eu2+ ions should correspond to a higher-energy (shorter wavelength) emission peak, while a lower-energy (longer wavelength) emission appears.21 Therefore, at y = 0.63, the Eu2+ ions mainly stay in the M1 sites, as confirmed by the similar d(M1–O) [2.59 Å, 440 nm] in the Mn series and d(M2–O) [2.60 Å, x = 0.15, 442 nm] in the Ba series.
To ensure high efficiency for the phosphor-converted WLED devices, a comprehensive understanding of the thermal quenching of phosphors is necessary.22–25 The relative emission intensity (IT/I0) for the Ba and Mn series from room temperature to 573 K are shown in Fig. 5a and b. The emission intensity decreases with the environmental temperature increase for all the samples because of the thermal quenching effect. However, the overall trend of thermal stability improvement by doping is evident in the Ba series (x = 0–1.59) and the Mn series (y = 0–0.63), with respect to the parent Sr2.97Eu0.03Y2(Si3O9)2 sample. In particular, the Ba series shows a clear thermal stability increase at the phase transition. The results can be governed by the increasing quenching activation barriers in both series (insert in Fig. 5b). Generally, excellent thermal stability could be expected in certain phosphors with high covalency, high rigidity and a small Stokes shift. The blue shifts in the Ba and Mn series cause the Stokes shift decrease, indicating a thermal energy increase (Ea). The Ea values of the Ba and Mn series were calculated by relation IT/I0 = [1 + Dexp(−Ea/kT)]−1, where IT (intensity at T), I0 (intensity at T = 0), D, and activation energy Ea are refined variables.10 Clearly, the Ea values of the Ba and Mn series gradually increase with x and y (insert in Fig. 5b), suggesting that the probability of nonradiative transition is weakened. Therefore, the thermal stability of the phosphors gradually increased on doping. A clear thermal quenching decrease appeared at the phase transition point, as shown by the dashed circle in Fig. 5a. This finding is also consistent with the Ea turning point during the phase transition (the insert in Fig. 5b). Thus, a jump change of thermal stability is possible in phosphor materials at phase transition. It is noted that the Mn series have a lower enhancement for the thermal stability than the Ba series, which can be attributed to the different thermal decay behavior between Eu2+ and Mn2+.
 | ||
| Fig. 5 Thermal quenching behaviour of photoluminescence for (a) Sr2.97−xBaxEu0.03Y2(Si3O9)2 (x = 0–1.59) and (b) Sr2.97−yMnyEu0.03Y2 (Si3O9)2 (y = 0–0.63) samples. Insert in (b) is the plot of activation energies dependent on x for Ba series and y for Mn series. | ||
To sum it up, the lattice-site control effect for Eu2+ ions discovered in M5(Si3O9)2 (M = Sr, Ba, Y, Mn) crystals can efficiently tune the photoluminescence and thermal quenching properties via the cation-substitution approach. This effect provides a new insight into the structural variations of the single-cation sites that are induced by phase transition and site-preferential occupancy, which are driven via size-mismatched cation substitution and can tune the luminescent properties of phosphor materials. This work reveals the mechanisms of optical adjustment by the coordination environment changing at specific sites. In particular, the abrupt structural change owing to phase transition offers unexpected and large-scale changes in optical properties. This effect can be extended to tune other properties, including the electric and magnetic properties that are sensitive to the structural variation at local sites.26–28
Acknowledgements
This project is financially supported by the National Natural Science Foundation of China (Grants No. NSFC 21301162, 21571162, 60977013, 91433110, U1301242, 21221061), the National College Students' Innovative Training Program (Nos. 201510491109, 201610491067, 201610491070), and the Ministry of Science and Technology of Taiwan (No. MOST 104-2917-I-564-060). Zewei Quan acknowledges the funding support (FRG-SUSTC1501A-17) from South University of Science and Technology of China.Notes and references
- N. C. George, K. A. Denault and R. Seshadri, Annu. Rev. Mater. Res., 2013, 43, 481–501 CrossRef CAS.
- R. J. Xie and N. Hirosaki, Sci. Technol. Adv. Mater., 2007, 8, 588–600 CrossRef CAS.
- P. Pust, V. Weiler, C. Hecht, A. Tücks, A. S. Wochnik, A.-K. Henß, D. Wiechert, C. Scheu, P. J. Schmidt and W. Schnick, Nat. Mater., 2014, 13, 891–896 CrossRef CAS PubMed.
- Z. G. Xia, C. G. Ma, M. S. Molokeev, Q. L. Liu, K. Rickert and K. R. Poeppelmeier, J. Am. Chem. Soc., 2015, 137, 12494–12497 CrossRef CAS PubMed.
- G. G. Li, Y. Tian, Y. Zhao and J. Lin, Chem. Soc. Rev., 2015, 44, 8688–8713 RSC.
- Y. Sato, H. Kato, M. Kobayashi, T. Masaki, D. H. Yoon and M. Kakihana, Angew. Chem., Int. Ed., 2014, 53, 7756–7759 CrossRef CAS PubMed.
- K. A. Denault, J. Brgoch, M. W. Gaultois, A. Mikhailovsky, R. Petry, H. Winkler, S. P. DenBaars and R. Seshadri, Chem. Mater., 2014, 26, 2275–2282 CrossRef CAS.
- Z. Y. Zhao, Z. G. Yang, Y. R. Shi, C. Wang, B. T. Liu, G. Zhu and Y. H. Wang, J. Mater. Chem. C, 2013, 1, 1407–1412 RSC.
- W. T. Chen, H. S. Sheu, R. S. Liu and J. P. Attfield, Inorg. Chem., 2012, 51, 8802–8809 CrossRef PubMed.
- S. S. Wang, W. T. Chen, Y. Li, J. Wang, H. S. Sheu and R. S. Liu, J. Am. Chem. Soc., 2012, 134, 8022–8025 CrossRef PubMed.
- F. W. Kang, H. S. Zhang, L. Wondraczek, X. B. Yang, Y. Zhang, D. Y. Lei and M. Y. Peng, Chem. Mater., 2016, 28, 2692–2703 CrossRef CAS.
- Z. G. Xia, G. K. Liu, J. G. Wen, Z. G. Mei, M. Balasubramanian, M. S. Molokeev, L. C. Peng, L. Gu, D. J. Miller, Q. L. Liu and K. R. Poeppelmeier, J. Am. Chem. Soc., 2016, 138, 1158–1161 CrossRef CAS PubMed.
- A. P. Tyutyunnik, I. I. Leonidov, L. L. Surat, I. F. Berger and V. G. Zubkov, J. Solid State Chem., 2013, 197, 447–455 CrossRef CAS.
- H. Yamane, T. Nagasawa, M. Shimada and T. Endo, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53, 1533–1536 Search PubMed.
- P. P. Dai, C. Li, X. T. Zhang, J. Xu, X. Chen, X. L. Wang, Y. Jia, X. J. Wang and Y. C. Liu, Light: Sci. Appl., 2016, 5, e16024 CrossRef CAS.
- M. Seibald, T. Rosenthal, O. Oeckler and W. Schnick, Crit. Rev. Solid State Mater. Sci., 2014, 39, 215–229 CrossRef CAS.
- W. B. Park, S. P. Singh and K. S. Sohn, J. Am. Chem. Soc., 2014, 136, 2363–2373 CrossRef CAS PubMed.
- G. K. Liu, Chem. Soc. Rev., 2015, 44, 1635–1652 RSC.
- W. B. Im, Y. Kim, H. S. Yoo and D. Y. Jeon, Inorg. Chem., 2009, 48, 557 CrossRef CAS PubMed.
- H. Terraschke and C. Wickleder, Chem. Rev., 2015, 115, 11352 CrossRef CAS PubMed.
- P. Dorenbos, ECS Solid State Lett., 2013, 2, R3001–R3011 CrossRef CAS.
- Z. J. Zhang, O. M. ten Kate, A. Delsing, E. van der Kolk, P. H. L. Notten, P. Dorenbos, J. T. Zhao and H. T. Hintzen, J. Mater. Chem., 2012, 22, 9813–9820 RSC.
- M. Y. Peng, X. W. Yin, P. A. Tanner, M. G. Brik and P. F. Li, Chem. Mater., 2015, 27, 2938–2945 CrossRef CAS.
- C. R. Ronda, Luminescence: From Theory to Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008 Search PubMed.
- Y. Q. Li, N. Hirosaki, R. J. Xie, T. Takeda and M. Mitomo, Chem. Mater., 2008, 20, 6704–6714 CrossRef CAS.
- S. Zhang, T. Saito, M. Mizumaki, W. T. Chen, T. Tohyama and Y. Shimakawa, J. Am. Chem. Soc., 2013, 135, 6056–6060 CrossRef CAS PubMed.
- W. Zheng, P. Huang, D. T. Tu, E. Ma, H. M. Zhu and X. Y. Chen, Chem. Soc. Rev., 2015, 44, 1379–1415 RSC.
- F. W. Kang, X. B. Yang, M. Y. Peng, L. Wondraczek, Z. J. Ma, Q. Y. Zhang and J. R. Qiu, J. Phys. Chem. C, 2014, 118, 7515–7522 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, Rietveld structural refinement processing and parameters, XRD patterns, Raman spectra, PLE and PL spectra, CIE color coordinates, including Fig. S1–S27 and Tables S1–S3, for Sr2.97−xBaxEu0.03Y2(Si3O9)2 (x = 0–1.59) and Sr2.97−yMnyEu0.03Y2(Si3O9)2 (y = 0–0.63) compounds. See DOI: 10.1039/c6ra11681g |
| ‡ These authors contribution to this work are equal. |
| This journal is © The Royal Society of Chemistry 2016 |