
Zeolite Y nanoparticle assemblies with high activity in the direct hydration of terminal alkynes†
Shuling Xuab,
Zhi Yun*a,
Yu Fengb,
Ting Tangb,
Zhongxue Fangb and
Tiandi Tang*b
aCollege of Chemistry and Chemical Engineering, Nanjing Tech University, Nanjing, Jiangsu 210009, P. R. China. E-mail: yunzhi@njtech.edu.cn
bSchool of Petrochemical Engineering, Changzhou University, Changzhou, Jiangsu 213164, P. R. China. E-mail: tangtiandi@wzu.edu.cn
First published on 7th July 2016
Abstract
A strong acidic zeolite Y nanoparticle assembly (HNANO-Y) with a micro–meso–macroporous structure was synthesized and used as a highly efficient heterogeneous catalyst for the hydration of alkynes to prepare ketone compounds, as compared to acidic mesoporous zeolite ZSM-5 and Beta catalysts. This feature should be assigned to the fact that the micro–meso–macroporous structure in the HNANO-Y benefits mass transfer and the strongly acidic sites on HNANO-Y facilitate alkyne hydration activity. The catalyst can be reused six times without loss of activity.
1. Introduction
Catalytic hydration of C–C triple bond provides a perfect and environmentally benign approach for the preparation of carbonyl compounds with 100% atomic efficiency.1–3 For instance, ketones are important structural motifs in modern organic synthesis that can be efficiently synthesized by Markovnikov hydration of alkynes over Hg(II) salts combining with acids in industrial processes.4–7 However, these catalytic systems always suffer from the high toxicity of mercury. Although many transition metal catalysts such as Ru,8,9 Rh,10,11 Pd,12,13 Pt,14,15 Ag,16,17 Au,18–20 Fe,21,22 Co23,24 and Ir25 have been widely attempted for this reaction process, these catalytic systems suffered from lower reactivity, recovery and reusage of the catalysts. Despite the concentrated solution of strong acids being extensively investigated, and exhibiting high activity for promotion of the alkyne hydration process,26–28 these catalytic systems always needed a large excess amount of acidic reagents and are unsuitable industrial applications.To solve these problems, in recent years, recyclable and recoverable heterogeneous catalysts, such as polystyrene supported sulfonic acid, Au modified mesoporous silica, silver exchanged silicotungstic acid (AgSTA) and Sn–W mixed oxide have been used for this process.29–31 Although these catalysts showed high efficiency, mineral acid was required as a co-catalyst. More recently, conventional micropore zeolites such as Beta, ZSM-5 and MOR as acidic catalysts were applied for these alkyne hydration reactions.32 However, the main drawback is that the relatively small pores in the zeolite crystals strongly limited molecular diffusion and bulk molecule conversion. As continuous work focuses on developing high efficient heterogeneous catalysts for organic synthesis,33–37 in this work, we synthesized for the first time nanoparticle assemblies of zeolite Y (NANO-Y) that presents good crystallinity and has a large external surface area by addition of the silane ([(CH3O)3SiC3H6N(C2H5)2C16H33]Cl) into the zeolite Y synthesis gel. Meanwhile, the microporous zeolite Y, and mesoporous zeolites ZSM-5 and Beta (ZSM-5-M and Beta-M) were also synthesized. After transforming zeolites to the H-form by ion-exchange in NH4NO3 solution, the obtained samples were denoted as HNANO-Y, HY, HZSM-5-M and HBeta-M. Compared with HY, HZSM-5-M and HBeta-M, HNANO-Y exhibits high activity in alkyne hydration reactions.
2. Experimental
2.1 Material synthesis
Zeolite Y nanoparticle assemblies (NANO-Y) were synthesized from an aluminosilicate gel with molar ratios of 1.0Al2O3/4.4Na2O/9.8SiO2/0.8TMOAC/153H2O, where TMOAC is a silane of N,N-diethyl-N-cetyl-N-(3-trimethoxysilylpropyl) ammonium chloride ([(CH3O)3SiC3H6N(C2H5)2C16H33]Cl). In a typical run, 18.7 mL of waterglass, 3.0 mL of H2O, and 2.81 mL of zeolite Y seed solution (1.409 g NaAlO2, 12.7 mL H2O, 5.12 g NaOH and 16.8 mL waterglass were mixed and aged for 24–36 h) were mixed. After further stirring for 180 min, 5.75 mL of an Al2(SO4)3 aqueous solution (45.6 wt%), and 5.95 mL aqueous solution of NaAlO2 (18.2 wt%) and NaOH (9.2 wt%) were added. After stirring at room temperature for 120 min, the obtained aluminosilicate gel was transferred into a Teflon-coated stainless-steel autoclave for static crystallization at 90 °C for 2 days. After filtrating and washing, the sample was dried at 120 °C overnight and calcined in air at 560 °C for 4 h. The microporous zeolite Y was also synthesized by similar procedures in the absence of the TMOAC. The mesoporous zeolite ZSM-5 and Beta (ZSM-5-M and Beta-M) were synthesized according to previous work procedures.38,39 The mesoporous-free Beta and ZSM-5 were prepared according to the procedure reported in our precious work.37,38 The H-form of the zeolite samples were obtained by ion-exchanging twice with NH4NO3 solution (1 M) at 80 °C for 4 h, followed by calcination at 550 °C for 4 h, and were denoted as HNANO-Y, HY, HZSM-5-M, HBeta-M, HBeta and HZSM-5.2.2 Catalyst characterization
X-ray powder diffraction (XRD) analysis was conducted on a Rigaku Smart Lab diffractometer, using Cu Kα radiation. Nitrogen adsorption–desorption isotherms were obtained using a Micromeritics ASAP2020M apparatus at the temperature of liquid nitrogen (−196 °C). Specific surface areas were calculated from the adsorption data, using the Brunauer–Emmett–Teller (BET) equation. The pore size distributions were calculated using the Barrett–Joyner–Halenda (BJH) model. Scanning electron microscopy (SEM) images were obtained on a NanoSEM200 apparatus. Transmission electron microscopy (TEM) images of the sample were obtained on a JEM-2100 microscope with a limited line resolution capacity of 1.4 Å, under a voltage of 200 kV. The acidity of the samples was measured using temperature-programmed desorption of ammonia (NH3-TPD) on a Micromeritics ASAP 2920 instrument. About 200 mg of sample was placed in a quartz tubular reactor and pretreated in a helium stream, heated to 450 °C with a rate of 1 °C min−1, and kept at 450 °C for 2 h. After the sample was cooled to 120 °C, NH3–He mixed gas (10 vol% NH3) was passed over the sample for 30 min. After removal of physically adsorbed NH3 by flowing helium for 2 h at 120 °C, the sample was heated from 120 to 500 °C with a rate of 10 °C min−1. The desorbed NH3 was collected in dilute hydrochloric acid and titrated with a dilute sodium hydroxide solution to determine the acidic site density of the sample.2.3 Reaction and analysis
All reagents, unless otherwise noted, were obtained from commercial sources (purity > 99%) and used without further purification. The typical experimental procedure for the alkyne hydration reaction was as follows: 30 mg catalyst, 0.5 mmol alkynes, 1.5 mL water were added into a 10 mL glass vessel. The reaction proceeded at the setting temperature for the desired time. After the reaction was finished, the catalyst was separated by centrifugation and filtration to obtain the liquid phase. The liquid products were analyzed by an Agilent 7890B gas chromatograph equipped with a flame ionization detector. The pure product was obtained by flash column chromatography on silica gel using petroleum ether (60–90 °C) and ethyl acetate as eluents. Compounds described in the literature were characterized by comparing their 1H and 13C NMR spectra to the reported data. 1H and 13C NMR spectra were recorded with a Bruker Avance 400 spectrometer at 400 and 100 MHz, respectively. Chemical shifts (δ) and coupling constants (J) are given in ppm and Hz, respectively. The chemical shifts are recorded relative to the residual solvent as an internal standard ([D1] chloroform: δ = 7.26 ppm for 1H and 77.0 ppm for 13C).The scale-up experiment was carried out in an autoclave (Parr 5500, 300 mL). The reaction mixture was as follows: 3 g HNANO-Y catalyst, 50 mmol 1-ethynyl-4-methoxybenzene and 150 mL water were placed into an autoclave. The reaction temperature was 80 °C, reaction time was 12 h, and stirring rate was 500 rpm. For the catalyst recycling experiment, once the reaction was finished, the solid catalyst was separated by centrifugation, and washed with 5 mL of acetone. The washing/centrifugation sequence was repeated three additional times. Finally, the obtained catalyst was dried under vacuum and reused for the next run.
3. Results and discussion
3.1 Catalyst characterization
Fig. 1a shows the XRD pattern and nitrogen sorption isotherm of the calcined NANO-Y sample. The XRD pattern shows that well-resolved peaks in the range of 5–50°, with a zeolite FAU structure, can be clearly observed in NANO-Y and Y samples, indicating the high degree of crystallinity of the two samples. The nitrogen adsorption–desorption isotherms of the NANO-Y exhibit a hysteresis loop at a relative pressure of 0.5–0.95, which is typically assigned to the presence of the mesoporous (Fig. 1b). Correspondingly, the mesopore-size diameter is mainly centered at 29.0 nm (inset, Fig. 1b). The sample texture parameters are shown in Table 1. HNANO-Y exhibits high BET and external surface areas and mesoporous volume (811 and 158 m2 g−1, and 0.22 cm3 g−1), which could benefit the mass transfer and provide abundant accessible acidic active sites.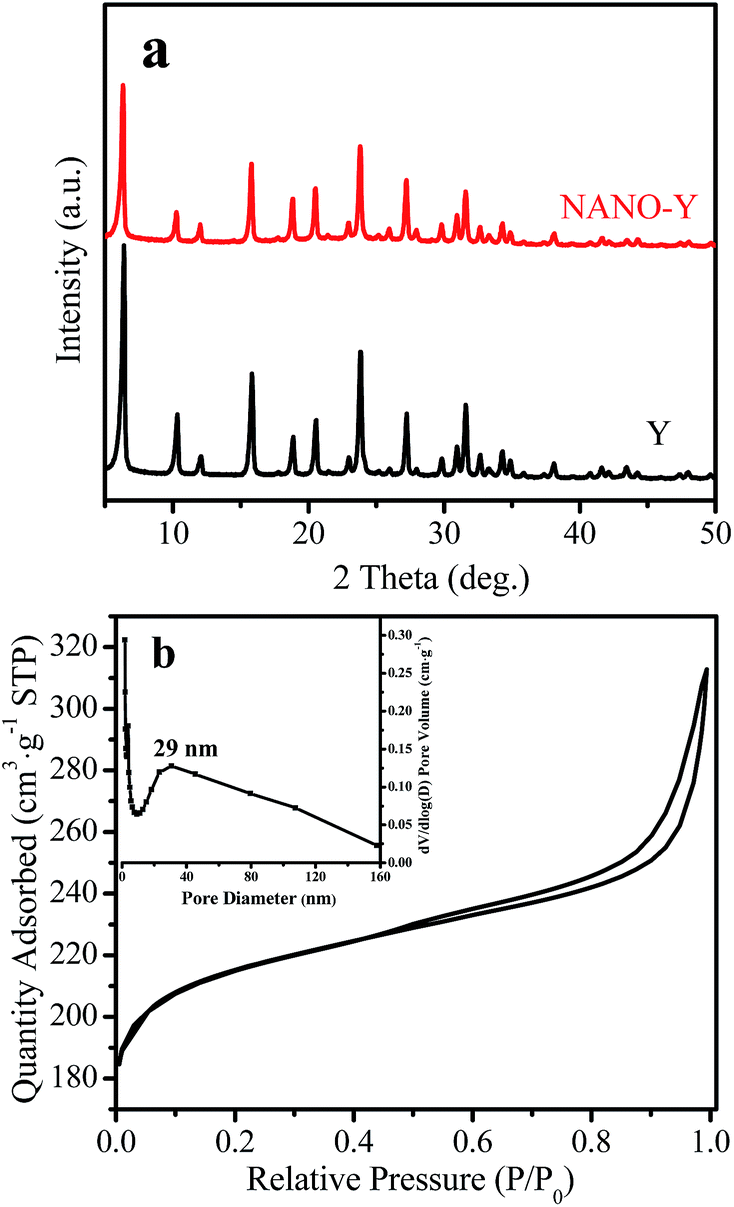 | ||
| Fig. 1 (a) XRD patterns of the Y and NANO-Y samples, and (b) N2 adsorption isotherms of the NANO-Y (inset, mesoporous size distribution of the NANO-Y sample). | ||
| Samples | SBETa (m2 g−1) | Vmicb (cm3 g−1) | Sextc (m2 g−1) | Smicd (m2 g−1) | Vmese (cm3 g−1) |
|---|---|---|---|---|---|
| a BET surface area.b Microporous volume.c External surface area, including mesoporous surface area.d Microporous surface area.e Mesoporous volume. | |||||
| HNANO-Y | 811 | 0.26 | 158 | 653 | 0.22 |
| HY | 821 | 0.32 | 59 | 762 | 0.06 |
| HZSM-5-M | 434 | 0.08 | 167 | 267 | 0.47 |
| HBeta-M | 619 | 0.18 | 180 | 439 | 0.29 |
| HBeta | 600 | 0.16 | 55 | 535 | 0.04 |
| HZSM-5 | 376 | 0.13 | 23 | 353 | 0.02 |
The SEM images show the zeolite Y nanoparticle assemblies with a particle size of 0.5–1.5 μm (Fig. 2a). The high resolution image shows that the particle is composed of nanoparticle aggregates with a main size of 60–100 nm (Fig. 2b). In this manner, the interstice mesopores and macropores could be formed in aggregates. The TEM image shows the Y nanoparticles with a highly crystalline micropore structure (Fig. 2c). The SEM, TEM, and nitrogen sorption results indicate that the NANO-Y exhibits a micro–meso–macroporous structure, which could benefit mass transfer and improve its catalytic performance. The NH3-TPD curves show that the weak (120–300 °C) and strong (300–450 °C) acidic sites existed on the HNANO-Y, HY, HZSM-5-M and HBeta-M samples (Fig. 3). However, not only the weak acidic sites, but also the strong acidic sites on the HNANO-Y and HY samples are much larger than those on the HZSM-5-M and HBeta-M samples. The acid–base titration results also show that the total acidity of the HNANO-Y is 830 μmol g−1, similar to HY (798 μmol g−1), and much higher than that of HBeta-M (256 μmol g−1) and HZSM-5-M (350 μmol g−1) samples (Table S1†).
 | ||
| Fig. 2 SEM images of the NANO-Y in the low (a) and high resolution (b), and (c) TEM image of the NANO-Y sample. | ||
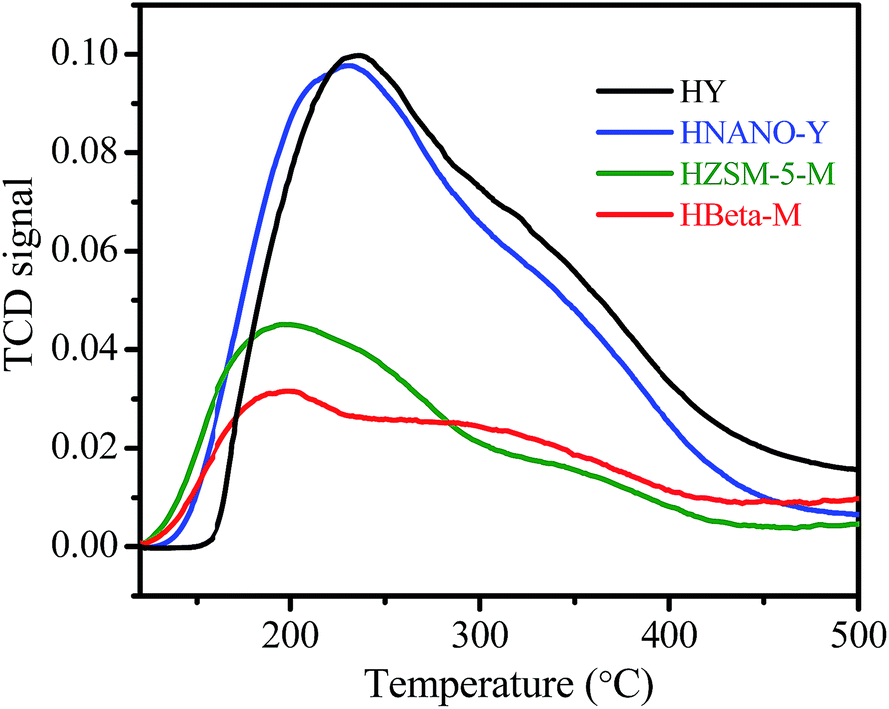 | ||
| Fig. 3 NH3-TPD curves of HY, HNANO-Y, HZSM-5-M and HBeta-M samples. | ||
3.2 Catalyst performance
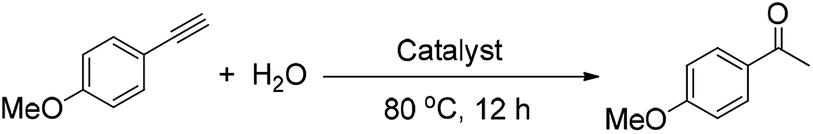
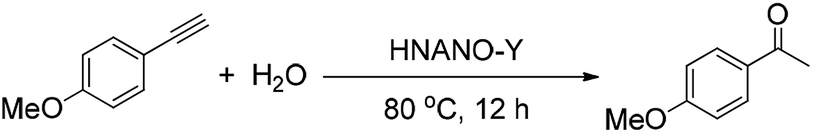
The hydration of alkynes to synthesize ketone compounds was firstly studied by choosing 1-ethynyl-4-methoxybenzene as a model substrate to investigate the activity of the series of catalysts (Table 2). To our delight, the highly desired product yield (99%) is obtained over the HNANO-Y catalyst (entry 2). The micropore HY catalyst gives substrate conversion of 71% (entry 3), much lower than that of HNANO-Y. It should be noted that the product is indeed formed over the HZSM-5-M and HBeta-M catalysts with good substrate conversions (entries 4 and 5). However, mesopore-free HZSM-5 and HBeta give low substrate conversions of 53 and 61%, respectively (entries 6 and 7). In addition, when the reaction was scaled-up to 100 times using the HNANO-Y catalyst, the obtained product yield is 96% (entry 8, Table 2). The water amount in the reaction mixture on the reaction activity was also surveyed, and the results are shown in the Table S2.† The conversion of 1-ethynyl-4-methoxybenzene is decreased when the mole ratio of water with reaction substrate is lower than 4. The substrate conversion on reaction time over HNANO-Y, HZSM-5-M and HBeta-M catalysts is shown in Fig. 4. Obviously, the substrate conversion over HNANO-Y is higher than that over HBeta-M and HZSM-5-M at the same reaction time. Under all conversions, the product selectivity is close to 100% on the HNANO-Y, HBeta-M and HZSM-5-M catalysts.
|
|||
|---|---|---|---|
| Entry | Catalyst | Conversion (%) | Selectivity (%) |
| a Reaction conditions: 1-ethynyl-4-methoxybenzene (0.5 mmol), water (1.5 mL), catalyst (30 mg), 80 °C for 12 h.b The reaction scaled-up 100 times of entry 2 in an autoclave. | |||
| 1 | None | — | — |
| 2 | HNANO-Y | 99 | 100 |
| 3 | HY | 71 | 100 |
| 4 | HZSM-5-M | 81 | 100 |
| 5 | HBeta-M | 88 | 100 |
| 6 | HZSM-5 | 53 | 100 |
| 7 | HBeta | 61 | 100 |
| 8 | HNANO-Yb | 96 | 100 |
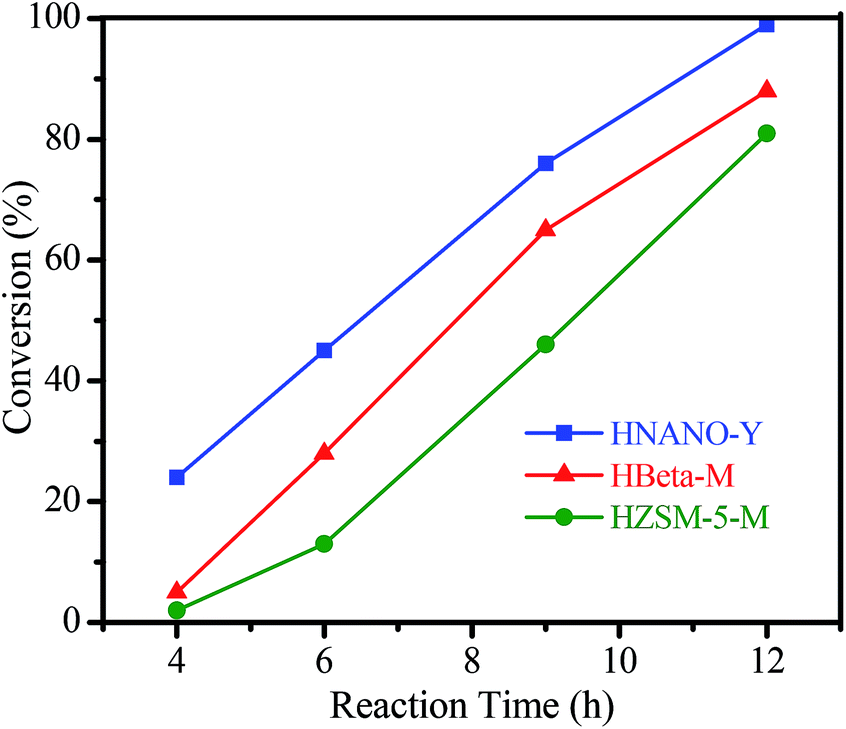 | ||
| Fig. 4 Dependence of the 1-ethynyl-4-methoxybenzene conversion on reaction time over HNANO-Y, HBeta-M and HZSM-5-M catalysts (0.5 mmol 1-ethynyl-4-methoxybenzene, 1.5 mL water, 30 mg HNANO-Y, the reaction temperature is 80 °C). | ||
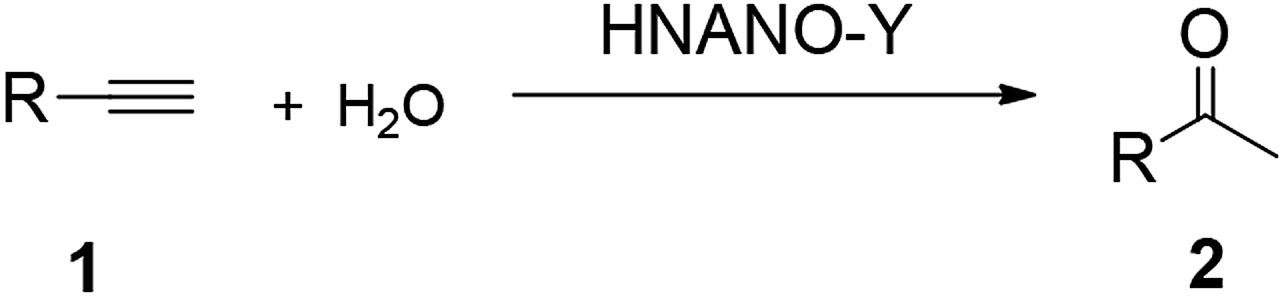
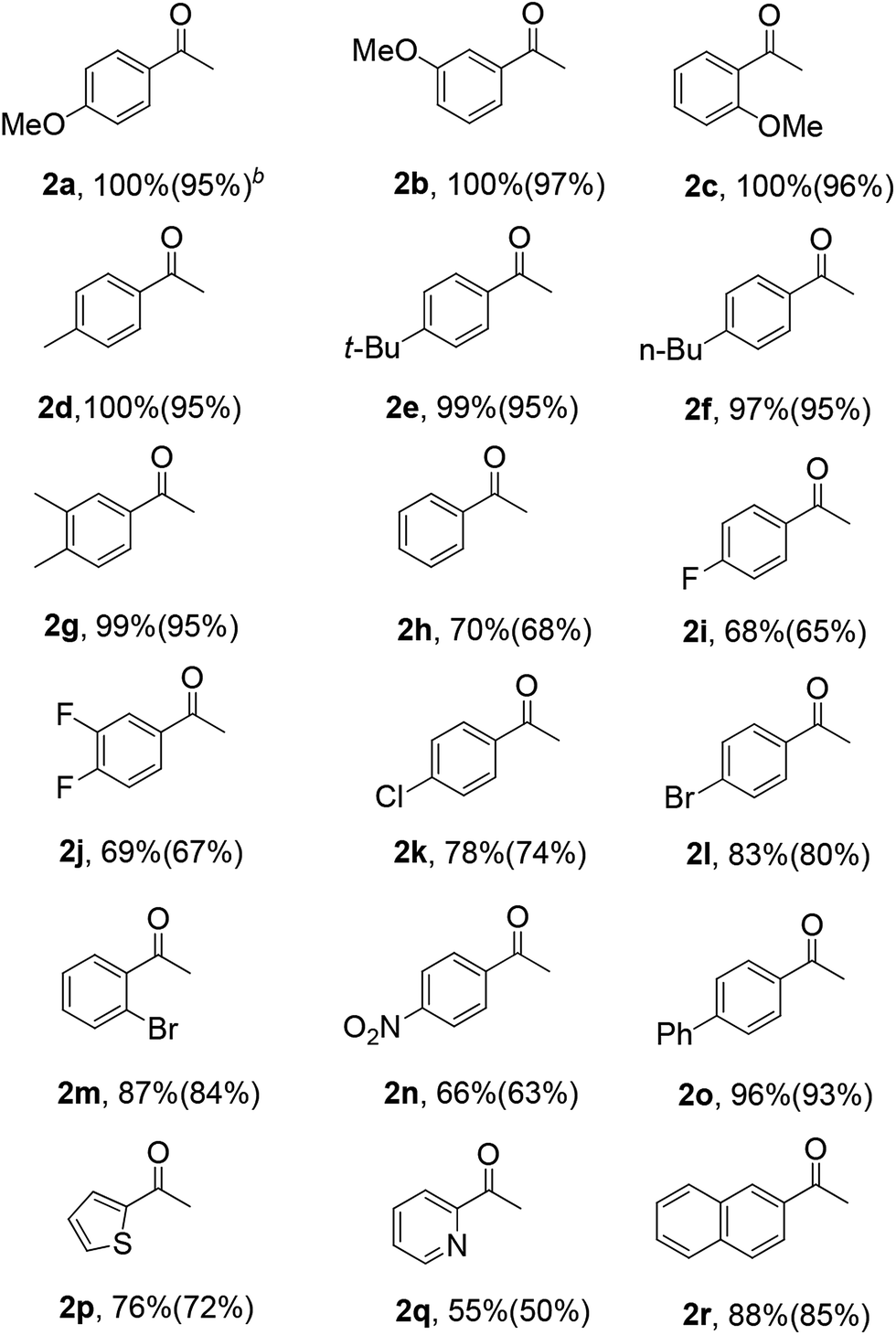
Table 3 shows the hydration activity of the HNANO-Y catalyst for a series of alkynes. The reaction exhibits good tolerance toward various aromatic alkynes with electron-donating and electron-withdrawing groups, such as methoxy, alkyl, halogen, nitro and phenyl at the ortho, meta or para positions, giving the corresponding products in moderate to excellent yields (2a–2o, 66–100%). Meanwhile, the alkynes bearing 2-thienyl and 2-pyridyl groups are compatible for this transformation (2p and 2q). Furthermore, the sterically hindered naphthyl-substituted alkynes also yielded the target product 2r in 88% yield.
Following this, to get further insights, an isotope labeling experiment was also conducted. The reaction proceeded using deuterium oxide instead of water under the standard conditions, giving deuterated product [D]-2a′ in 98% yield (eqn (1)). This result demonstrates that the two hydrogen atoms of product 2a at the methyl position stem from water.
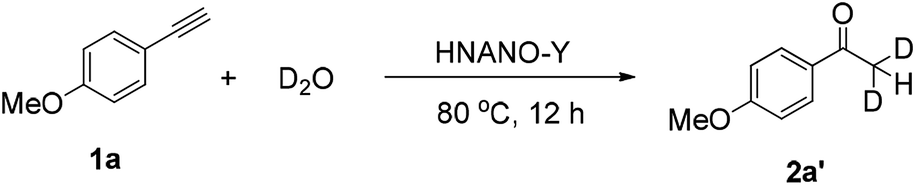 | (1) |
The reusability of the HNANO-Y catalyst also served in the synthesis of 4-methoxyacetophenone from 1-ethynyl-4-methoxybenzene and the results are presented in Table 4. The catalyst recycling experimental results show that the catalyst is still efficient at promoting this process even after six runs without significant loss in catalytic activity.
|
||
|---|---|---|
| Entry | Recycle | Conversion (%) |
| a Reaction conditions: 1-ethynyl-4-methoxybenzene (0.5 mmol), water (1.5 mL), HNANO-Y (30 mg) at 80 °C for 12 h. | ||
| 1 | Run 1 | 99 |
| 2 | Run 2 | 99 |
| 3 | Run 3 | 98 |
| 4 | Run 4 | 98 |
| 5 | Run 5 | 97 |
| 6 | Run 6 | 97 |
3.3 Discussion on the high activity
The high activity and broad scope of reactants over the HNANO-Y catalyst should be attributed to the abundant acidic sites with strong acidity on the HNANO-Y, and especially the micro–meso–macroporous structure in the HNANO-Y. The HY and HANO-Y present similar acidity (Fig. 3) and BET surface area (Table 1), but the conversion of the 1-ethynyl-4-methoxybenzene over HNANO-Y (99%) is much higher than that over HY (71%) in the hydration reaction (Table 2). This is due to the presence of meso–macroporous structure in the HNANO-Y, which could favor mass transfer and improve its catalytic performance. It is mentioned that the mesopore size of HBeta-M and HZSM-5-M are mainly centered at 10.5 and 21.8 nm (Fig. S1†), and the mesoporous size of NANO-Y is centered at 29 nm (Fig. 1b inset). In addition, compared with HNANO-Y (158 m2 g−1, 0.22 cm3 g−1), the HZSM-5-M and HBeta-M show large mesoporous surface areas (167 and 180 m2 g−1, respectively) and mesoporous volume (0.47 and 0.29 cm3 g−1, respectively, Table 1), but the 1-ethynyl-4-methoxybenzene conversion over HNANO-Y (99%) is higher than over HZSM-5-M (81%) and HBeta-M (88%) catalysts (Table 2). This can be interpreted as follows: not only are the acidic sites on the HANO-Y higher than on the HZSM-5-M and HBeta-M, but the acidic strength of the HNANO-Y is also stronger than HZSM-5-M and HBeta-M (Fig. 3), which could enhance the reaction activity. Furthermore, HNANO-Y has relatively large micropores (0.74 nm) and high micropore surface areas (653 m2 g−1),39 the acidic sites in the micropores could be easily accessible for 1-ethynyl-4-methoxybenzene. In the contrast, HZSM-5-M and HBeta-M have relatively small micropores (0.53 × 0.56 nm and 0.66 × 0.77 nm, respectively) and low micropore surface areas (353 and 535 m2 g−1, respectively),39 which disfavor 1-ethynyl-4-methoxybenzene molecule diffusion into the micropore channels.4. Conclusions
In summary, a micro–meso–macroporous zeolite Y nanoparticle assembly (HNANO-Y) was synthesized and applied to hydration of alkynes for the synthesis of ketone compounds. Compared with mesoporous zeolite ZSM-5 and Beta (HZSM-5-M and HBeta-M) catalysts, HNANO-Y shows high activity and good compatibility with a broad range of functional groups. This feature could be attributed to the abundant acidic sites on the HNANO-Y that can highly effectively catalyze the alkyne hydration reaction. In addition, the meso–macroporous in the HNANO-Y could facilitate the mass transfer and further improve its catalytic performance. The catalyst could be recycled up to six consecutive cycles without loss in activity.Acknowledgements
This study was supported by the National Natural Science Foundation of China (21506017 and 21476030) and the Natural Science Foundation of Jiangsu Province of China (BK20150258).References
- L. Hintermann and A. Labonne, Synthesis, 2007, 1121 CrossRef CAS.
- F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 14, 3079 CrossRef PubMed.
- M. Beller, J. Seayad, A. Tillack and H. Jiao, Angew. Chem., Int. Ed., 2004, 43, 3368 CrossRef CAS PubMed.
- M. Kutscheroff, Chem. Ber., 1881, 1540 CrossRef.
- B. Neumann and H. Schneider, Angew. Chem., Int. Ed., 1920, 33, 189 CrossRef CAS.
- D. A. Ponomarev and S. M. Shevchenko, J. Chem. Educ., 2007, 84, 1725 CrossRef CAS.
- W. L. Budde and R. E. Dessy, J. Am. Chem. Soc., 1963, 85, 3964 CrossRef CAS.
- J. Halpern, B. R. James and A. L. W. Kemp, J. Am. Chem. Soc., 1961, 63, 4097 CrossRef.
- M. M. Taqui Khan, S. B. Halligudi and S. Shukla, J. Mol. Catal., 1990, 58, 299 CrossRef.
- B. R. James and G. L. Rempel, J. Am. Chem. Soc., 1969, 91, 863 CrossRef CAS.
- X. Liu, L. Liu, Z. Wang and X. Fu, Chem. Commun., 2015, 51, 11896 RSC.
- Y. Fukuda, H. Shiragami, K. Utimoto and H. Nozaki, J. Org. Chem., 1991, 56, 5816 CrossRef CAS.
- C. Xu, W. Du, Y. Zeng, B. Dai and H. Guo, Org. Lett., 2014, 16, 948 CrossRef CAS PubMed.
- W. Hiscox and P. W. Jennings, Organometallics, 1990, 9, 1997 CrossRef CAS.
- L. W. Francisco, D. A. Moreno and J. D. Atwood, Organometallics, 2001, 20, 4237 CrossRef CAS.
- M. B. T. Thuong, A. Mann and A. Wagner, Chem. Commun., 2012, 48, 434 RSC.
- K. T. Venkateswara, P. S. S. Prasad and N. Lingaiah, Green Chem., 2012, 14, 1507 RSC.
- A. Cavarzan, A. Scarso, P. Sgarbossa, G. Strukul and J. N. H. Reek, J. Am. Chem. Soc., 2011, 133, 2848 CrossRef CAS PubMed.
- L. Li and S. B. Herzon, J. Am. Chem. Soc., 2012, 134, 17376–17379 CrossRef CAS PubMed.
- S. G. Weber, D. Zahner, F. Rominger and B. F. Straub, ChemCatChem, 2013, 5, 2330 CrossRef CAS.
- X. F. Wu, D. Bezier and C. Darcel, Adv. Synth. Catal., 2009, 351, 367 CrossRef CAS.
- J. Park, J. Yeon, P. H. Lee and K. Lee, Tetrahedron Lett., 2013, 54, 4414 CrossRef CAS.
- T. Tachinami, T. Nishimura, R. Ushimaru, R. Noyori and H. Naka, J. Am. Chem. Soc., 2013, 135, 50 CrossRef CAS PubMed.
- S. Wang, C. Miao, W. Wang, Z. Lei and W. Sun, ChemCatChem, 2014, 6, 1612 CrossRef CAS.
- T. Hirabayashi, Y. Okimoto, A. Saito, M. Morita, S. Sakaguchi and Y. Ishii, Tetrahedron, 2006, 62, 2231 CrossRef CAS.
- A. D. Allen, Y. Chiang, A. J. Kresge and T. T. Tidwell, J. Org. Chem., 1982, 47, 775 CrossRef CAS.
- T. Tsuchimoto, T. Joya, E. Shirakawa and Y. Kawakami, Synlett, 2000, 12, 1777 Search PubMed.
- N. Olivi, E. Thomas, J. F. Peyrat, M. Alami and J. D. Brion, Synlett, 2004, 2175 CAS.
- J. Huang, F. Zhu, W. He, F. Zhang, W. Wang and H. Li, J. Am. Chem. Soc., 2010, 132, 1492 CrossRef CAS PubMed.
- X. Jin, T. Oishi, K. Yamaguchi and N. Mizuno, Chem.–Eur. J., 2011, 17, 1261 CrossRef CAS PubMed.
- K. T. V. Rao, P. S. S. Prasad and N. Lingaiah, Green Chem., 2012, 14, 1507 RSC.
- N. Mameda, S. Peraka, M. R. Marri, S. Kodumuri, D. Chevella, N. Gutta and N. Nama, Appl. Catal., A, 2015, 505, 213 CrossRef CAS.
- S. Chen, Z. Shao, Z. Fang, Q. Chen, T. Tang, W. Fu, L. Zhang and T. Tang, J. Catal., 2016, 338, 38 CrossRef CAS.
- W. Fu, T. Liu, Z. Fang, Y. Ma, X. Zheng, W. Wang, X. Ni, M. Hu and T. Tang, Chem. Commun., 2015, 51, 5890 RSC.
- W. Fu, Y. Feng, Z. Fang, Q. Chen, T. Tang, Q. Yu and T. Tang, Chem. Commun., 2016, 52, 3115 RSC.
- T. Liu, W. Fu, X. Zheng, J. Jiang, M. Hu and T. Tang, RSC Adv., 2014, 4, 18217 RSC.
- X. Zheng, W. Fu, J. Xiong, J. Xi, X. Ni and T. Tang, Catal. Today, 2016, 264, 152 CrossRef CAS.
- W. Fu, L. Zhang, D. Wu, M. Xiang, Q. Zhuo, K. Huang, Z. Tao and T. Tang, J. Catal., 2015, 330, 423 CrossRef CAS.
- W. Fu, L. Zhang, T. Tang, Q. Ke, S. Wang, J. Hu, G. Fang, J. Li and F.-S. Xiao, J. Am. Chem. Soc., 2011, 133, 15346 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra11489j |
| This journal is © The Royal Society of Chemistry 2016 |