
New synthetic strategy targeting well-defined α,ω-telechelic polymethylenes with hetero bi-/tri-functionalities via polyhomologation of ylides initiated by new organic boranes based on catecholborane and post functionalization
Abstract
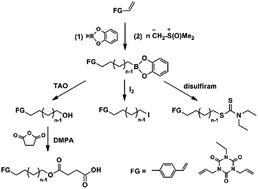
A series of well-defined α,ω-telechelic polymethylenes with hetero bi-/tri-functionalities were synthesized by a tandem strategy combining polyhomologation of ylides initiated by new organic boranes based on catecholborane with post functionalization using end-capping reagents and esterification. The chain structures of such polymers were attested by 1H NMR spectra, FT-IR and GPC. The functionality can reach up to 100%, and the molecular weights of the obtained polymers showed a range from 870 to 12 080 g mol−1 with a narrow distribution (Đ = 1.04–1.12).
 Please wait while we load your content...
Please wait while we load your content...

