
Sensitive and selective detection of Hg2+ based on an electrochemical platform of PDDA functionalized rGO and glutaraldehyde cross-linked chitosan composite film

Abstract
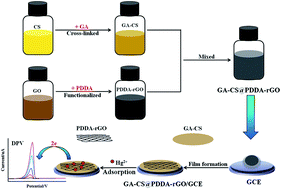
In this paper, a uniform PDDA-functionalized graphene composite film (GA–CS@PDDA-rGO) was utilized for detection of trace Hg2+ by using glutaraldehyde cross-linked chitosan (GA–CS) as a Hg2+-chelating adsorbent and film-forming agent. The results showed that a well-defined and high-sensitivity stripping peak at 0.06 V for Hg2+ was observed at the GA–CS@PDDA-rGO/GCE. Moreover, two important affecting factors of the content of PDDA-rGO and deposition potential were optimized on the GA–CS@PDDA-rGO/GCE modified electrode. Under the optimal conditions, the GA–CS@PDDA-rGO/GCE modified electrode showed a good linearity in the range of 0.03–5 μM between the concentration of the Hg2+ and stripping peak current. The detection limit was estimated to be 7.7 nM (S/N = 3). The interference and selectivity of other heavy metal ions were evaluated, showing no obvious interference on the Hg2+ detection. The results indicated that the GA–CS@PDDA-rGO composite film provided an efficient strategy and a new promising platform for detection of Hg2+.
 Please wait while we load your content...
Please wait while we load your content...

