
Highly efficient non-doped blue electroluminescent materials for organic light-emitting devices†
Abstract
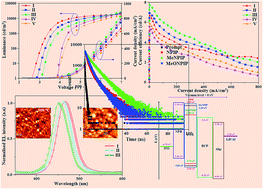
We have synthesized the new blue-light-emitting materials 4,4′-bis(1-(naphthalen-1-yl)-1H-phenanthro[9,10]imidazole-2-yl)-1,1′-biphenyl (NPIP), 4,4′-bis(4-methylnaphthalen-1-yl)-1H-phenanthro[9,10]imidazole-2-yl)-1,1′-biphenyl(MeNPIP) and 4,4′-bis(4-methoxynaphthalen-1-yl)-1H-phenanthro[9,10]imidazole-2-yl-1,1′-biphenyl(MeONPIP) through a two-step procedure using inexpensive catalysts. These compounds showed excellent thermal properties with a very high glass-transition temperature of 195–201 °C due to their rigid molecular backbones and they emit intense blue light in both solution and film. A device fabricated with MeONPIP shows maximum efficiencies (ηex 6.92%; ηc −7.80 cd A−1; ηp 6.10 lm W−1) with a low turn-on voltage and the performances of the device suggest that the phenanthroimidazole unit is an excellent building block for tuning carrier injection properties as well as for blue emission.
 Please wait while we load your content...
Please wait while we load your content...

