
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly stable supercapacitive performance of one-dimensional (1D) brookite TiO2 nanoneedles†
Rupesh S.
Devan
*abc,
Yuan-Ron
Ma
*c,
Ranjit A.
Patil
c and
Schmidt-Mende
Lukas
d
aCentre for Physical Sciences, School of Basic and Applied Sciences, Central University of Punjab, Bathinda, Punjab 151001, India. E-mail: devan_rs@yahoo.co.in
bDepartment of Physics, Savitribai Phule Pune University (Formerly, University of Pune), Pune 411007, India
cDepartment of Physics, National Dong Hwa University, Hualien 97401, Taiwan, Republic of China. E-mail: ronma@mail.ndhu.edu.tw
dDepartment of Physics, University of Konstanz, 78457 Constance, Germany
First published on 22nd June 2016
Abstract
We report the highly stable supercapacitive performance of one-dimensional (1D) nanoneedles of brookite (β) TiO2 synthesized on a conducting glass substrate. The 1D β-TiO2 nanoneedles synthesized over a large area array utilizing hot-filament metal vapor deposition (HFMVD) were ∼24–26 nm wide, ∼650 nm long and tapered in a downward direction. X-ray photoemission spectroscopy (XPS) revealed their chemical properties and stoichiometric Ti and O composition. The 1D β-TiO2 nanoneedles execute as parallel units for charge storage, yielding a specific capacitance of 34.1 mF g−1. Electrochemical impedance spectroscopy revealed that the large surface area and brookite crystalline nature of the 1D nanoneedles provided easy access to Na+ ions, and resulted in low diffusion resistance, playing a key role in their stable charging–discharging electrochemical mechanism. Moreover, the non-faradic mechanism of these nanoneedles delivered better durability and high stability up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, and a columbic efficiency of 98%. Therefore, 1D β-TiO2 nanoneedles hold potential as an electrode material for highly stable supercapacitive performance with long cycle lifetime.
000 cycles, and a columbic efficiency of 98%. Therefore, 1D β-TiO2 nanoneedles hold potential as an electrode material for highly stable supercapacitive performance with long cycle lifetime.
Introduction
Supercapacitors, known as electric double-layer capacitors, store charges at the interface of an electrolyte and an electrode of choice through reversible electrochemical diffusion of ions. With the increase in demand for high-performance energy storage technologies, supercapacitor electrode materials with fast recharging ability, high cycling stability, reversibility and long cycle lifetime are of importance. At present, supercapacitors provide already higher power density than batteries, and energy density than conventional capacitors. However, their stability and cycle lifetime are inferior to those of batteries and conventional capacitors. Carbon-based materials and conducting polymers known for high specific capacitance values and high power densities suffer from high energy densities even after achieving larger specific surface areas and rational porous morphology.1 Moreover, their swelling and shrinking during charging and discharging induces stress and results in short lifetime and faster degradation. Therefore, when it comes to the stability and long cycle lifetime, transition metal oxides seem to be a better alternative. Although the life cycle of transition metal oxides is limited from a few hundreds to a few thousand,2–4 efforts have been made for the improvement by reducing the structural dimensions of synthesized electrode materials. The nanoscale dimensions with well-defined geometry and colossal surface area are expected to improve the supercapacitive performance of the metal-oxides. Among all available morphologies of transition metal oxides, one-dimensional (1D) nanostructures plays an important role in the enhancement of charge storing ability, stability and cycle lifetime by providing large surface area and therefore more capillary pathways for ion diffusion/transport.5Over the past few decades, titanium dioxide (TiO2) is one of the most fascinating functional materials in the fabrication of supercapacitor because of the low processing cost, high chemical stability compared to most alternative materials, abundance and non-toxic nature. In the form of ultra-thin films or nanoparticles of size below 10 nm, mesoporous TiO2 films dramatically increase the ion transport and provide the high specific capacitance of 90–120 μF cm−2 at the normalized surface area.6 Highly ordered arrays of TiO2 nanotubes provide large surface area were found interesting alternative morphology for charge storage.3,7,8 However, the discontinuous and polycrystalline morphology of TiO2 nanotubes offers relatively poor conductivity and low electrochemical activity, limit its use in high-performance supercapacitors. Therefore, significant efforts have been made to synthesize TiO2 nanorods, nanowires, nanotubes and nanofibers, etc. and improve their supercapacitive performance in combination with various electrolytes such as H2SO4, NaOH, Na2SO4, LiClO4, and LiPF6, etc.3,5,7–11 Mainly three different polymorphs, rutile, anatase and brookite (β) are described regarding crystal structural arrangements of distorted TiO6 octahedra. Chains of edge-shared distorted octahedra running parallel to the c-axis yielding in β-phase of 1D nanoneedles. The shared edges of distorted TiO6 octahedra provide abundant enough vacant sites to accommodate Na+ ions. Therefore, especially β-TiO2 promises a electrode material for electrochemical processes. In nanocrystalline form, the β-phase is thermodynamically most stable with dimensions between 11 and 35 nm,12 and crystallographic data in brookite are intermediate between rutile and anatase phases.13 Moreover, β-TiO2 is promising dielectrics with much larger static dielectric constant14 and also known to exhibit markedly higher catalytic activities12 than anatase and rutile TiO2. Therefore, the β-phase is expected to offer high stability and long cycle lifetime.
So far, supercapacitor properties of anatase and rutile structures are studied extensively15–17 and seem to have reached their limit. Because of the difficulties encountered in the synthesis, most of the times β-phases was accompanied with anatase or/and rutile phases.12,18,19 Therefore, high temperature calcinations19 (700 °C) and annealing18 (800 °C) treatment was employed to obtain β-TiO2 nanorods. Nevertheless, the properties of the nanostructure of pure β-TiO2 phase seem to be interesting. To the best of our knowledge, supercapacitor properties of pure 1D β-TiO2 nanoneedles have not been reported yet in the literature. Therefore, in this paper, we report stable supercapacitive performance of 1D β-TiO2 nanoneedles synthesized in a large area via hot-filament metal vapor deposition (HFMVD) technique without additional heat treatment. The HFMVD technique is unique and useful for the synthesis of the variety of metal oxide nanostructures with diverse morphologies and crystalline phases.20–24 The structural morphology and chemical composition of the 1D β-TiO2 nanoneedles was examined utilizing field-emission scanning electron microscopy (FESEM) and X-ray photoemission spectroscopy (XPS), respectively. The electrochemical characterizations of the large-area array of the 1D β-TiO2 nanoneedles were carried out by cyclic voltammetry (CV), galvanostatic charge/discharge method, and AC impedance analysis in the electrolyte of Na2SO4. The results strongly indicated that 1D β-TiO2 nanoneedles provide columbic efficiency of 98%, high stability, greater retentivity (∼88.2%) and long cycle lifetimes up to 10000 cycles in Na2SO4 electrolyte. Therefore, the large area array of 1D β-TiO2 nanoneedles is an attractive material for fabrication of highly stable supercapacitors of long cycle lifetime.
Experimental
Large area arrays of TiO2 nanoneedles were synthesized utilizing hot-filament metal vapor deposition (HFMVD) technique. A clean titanium (Ti) wire (99.9% pure) with a diameter of 1 mm was passed through a pure graphite disc fixed on two supporting Cu electrodes mounted in a vacuum chamber. Once the pressure of the vacuum chamber was pumped down to 1 × 10−2 Torr, the Ti wire was heated to ∼1150 °C for 15 min to generate hot titanium vapor. The hot titanium vapor reacted with the residual oxygen (or leaking air) to form the metal oxide vapor of TiOx (x ≤ 2). When the TiOx vapor imparted the rather cold glass substrate coated with a conducting ITO thin film, it condensed into one-dimensional (1D) TiO2 nanoneedles. The surface morphology of the as-synthesized large-area array of TiO2 nanoneedles was characterized using field-emission scanning electron microscope (FESEM, JEOL JSM-6500F). The electronic structure and chemical state of these 1D TiO2 nanoneedles were analyzed using an X-ray photoelectron spectrometer (XPS, Thermo Scientific Inc. K-alpha) with a microfocus monochromated Al Kα X-ray. The 1D β-TiO2 nanoneedles based supercapacitor device constituted of the transparent 1D β-TiO2 nanoneedle array on a conducting ITO coated glass substrate as working electrode, a platinum foil as the counter electrode and saturated calomel electrode (SCE) stored in saturated KCl as the reference electrode was used to corroborate electrochemical properties. All the electrochemical measurements were performed with a potentiostat (Bio-Logic, SP-150) in 1 M Na2SO4 aqueous electrolyte at room temperature to investigate the potential of large area arrays of 1D TiO2 nanoneedles as a highly stable electrochemical supercapacitor. The cycling stability and performance of the device was confirmed with two terminal measurement method.Results and discussion
The FESEM images in Fig. 1 show the surface morphology of the large-area array of 1D TiO2 nanoneedles. In Fig. 1(a) and (b) a section of a large area array of 1D TiO2 nanoneedles is shown in the top and side view, respectively. The self-assembled 1D TiO2 nanoneedles are conical in shape and displays various diameters at the tip, body, and top. The array contains roughly 425 nanoneedles per square micrometer. The textural boundaries of these randomly dispersed TiO2 nanoneedles are clearly visible throughout the needle body. The high-magnification FESEM image in the inset of Fig. 1(a) shows single TiO2 nanoneedles. The conical shaped 1D TiO2 nanoneedles are on average ∼650 nm long and tapered in the downward direction, forming a film with an average thickness of ∼470 nm (Fig. 1(b)). The diameter of the nanoneedles is thinnest at the bottom (smaller than 12 nm) and ∼50 nm at the top for the thickest nanoneedles. However, a statistical histogram of the diameter at the top of the 1D TiO2 nanoneedles (Fig. 1(c)) clearly shows that the diameter at the top of most of the TiO2 nanoneedles falls in the range of 24 to 26 nm. The variation of the diameters of the TiO2 nanoneedles can be fitted with a log-normal distribution function as follows, | (1) |
![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) [≡26.82 ± 0.52 nm] is the mean (average) diameter of the various nanoneedles, A [≡111.41 ± 10.53] is the initial constant, and σ [≡0.19 ± 0.03] is the standard deviation of the diameters of the nanoneedles. The log-normal distribution of the nanoneedles diameters is asymmetrical. The small standard deviation of the diameter distribution (σ ≤ 0.25) indicates that the nanoneedles are well confined to a limited diameter range.
[≡26.82 ± 0.52 nm] is the mean (average) diameter of the various nanoneedles, A [≡111.41 ± 10.53] is the initial constant, and σ [≡0.19 ± 0.03] is the standard deviation of the diameters of the nanoneedles. The log-normal distribution of the nanoneedles diameters is asymmetrical. The small standard deviation of the diameter distribution (σ ≤ 0.25) indicates that the nanoneedles are well confined to a limited diameter range.
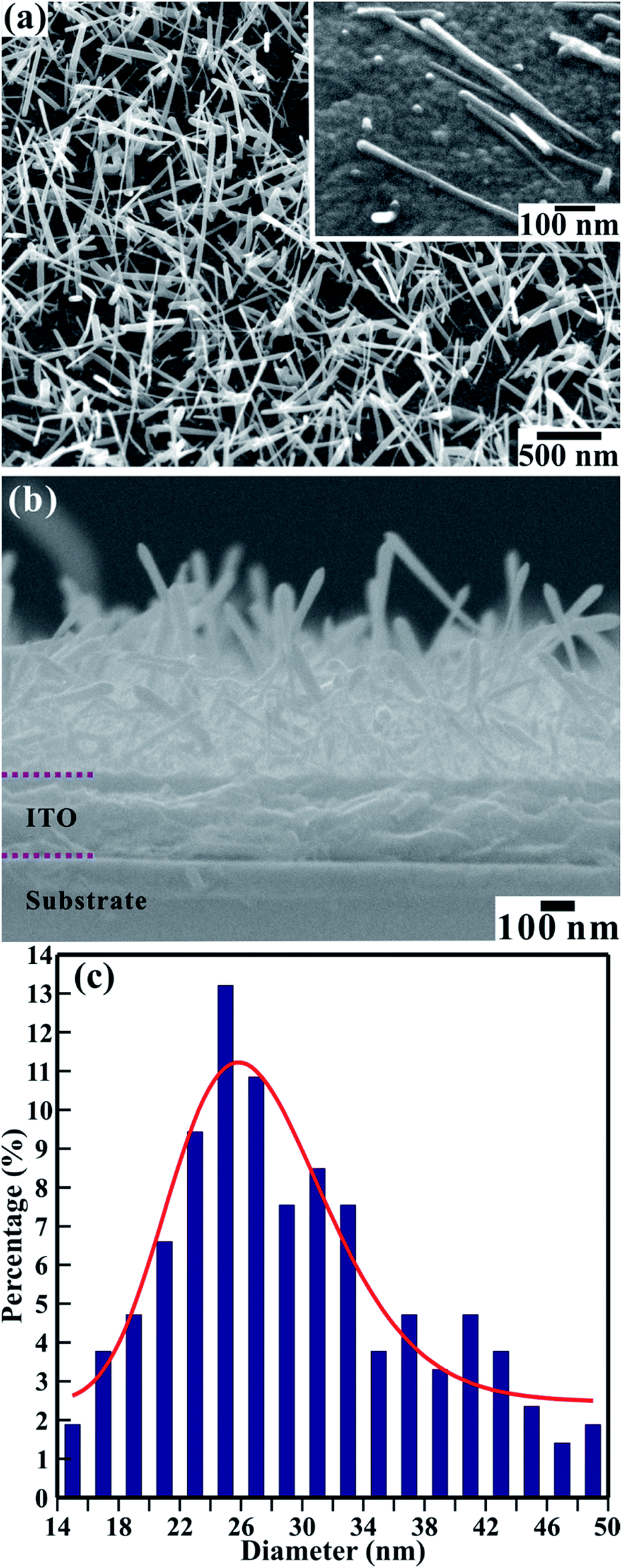 | ||
| Fig. 1 FESEM image showing the (a) top and (b) side views of the large-area array of 1D TiO2 nanoneedles on conducting glass substrate. Inset of (a) shows high-magnification FESEM image of the single 1D TiO2 nanoneedles separated from the array. (c) Statistical histogram of the distribution of diameter at the top of the TiO2 nanoneedles, fitted by a log-normal distribution function. | ||
Further, the crystalline structure of these well-defined TiO2 nanoneedles was confirmed by XRD and discussed elsewhere.25 The corresponding XRD spectra evidences that the 1D TiO2 nanoneedles are exclusively of orthorhombic crystal structure in brookite phase, which is assigned to the space group Pbca (JCPDS-761936) with lattice constants a = 0.919 nm, b = 0.546 nm, c = 0.516 nm and α = β = γ = 90°. Altogether, it demonstrates that our HFMVD technique can be successfully employed for the synthesis of 1D TiO2 nanostructures with optimal dimensions and required thickness. According to our literature survey, rutile and anatase phases are commonly synthesized and widely studied TiO2 phases. Only brookite is investigated rarely, even though it has exciting properties. Therefore, the large area array of 1D TiO2 nanoneedles of brookite phase reported here is of special importance in scientific and technological application point of view.
XPS was used to perform quantitative analysis of the electronic structure and chemical properties of the 1D β-TiO2 nanoneedles. Fig. 2 illustrate two Ti(2p) and O(1s) XPS spectra obtained for the large area array of 1D TiO2 nanoneedles. Fig. 2(a) shows the double peak features in the Ti(2p) XPS spectrum. To precisely determine the features of the double peaks of Ti(2p3/2) and Ti(2p1/2), the Ti(2p) XPS spectra was decomposed via Voigt curve fitting with the Shirley background. The deconvolution shows a perfect fit for two peaks located at binding energies of 458.56 and 464.24 eV, respectively, corresponding to the Ti(2p3/2) and Ti(2p1/2) core levels of Ti4+ cations and not of Ti3+.17,26 The energy separation between Ti(2p3/2) and Ti(2p1/2) peaks is 5.68 eV, and their area ratio is 2.48, which reflects a strong bonding between the Ti and O atoms. The full width at half-maximum (FWHM) of the Ti(2p3/2) and Ti(2p1/2) peak are 1.29, 2.27, respectively, indicating the high resolution of the Ti(2p) XPS spectrum in comparison with previous studies.26,27 Similarly, the O(1s) XPS spectrum of the 1D nanoneedles shown in Fig. 2(b) was decomposed via Voigt curve fitting with the Shirley background. The results demonstrate a perfect fit to two peaks located at 529.89 and 531.31 eV, with FWHM's of 1.38 and 2.19 eV, respectively. The lower binding energy peak at 530.14 eV corresponds to the O(1s) core level of the O2− anions in the 1D β-TiO2 nanoneedles. However, the higher binding peak at 531.61 eV is ascribed to surface contamination, such as carbon oxides or hydroxides20,21 of the 1D nanoneedles. The O(1s) peak observed at a binding energy of 529.89 eV is associated with the Ti–O chemical bonding.17 The atomic ratio of titanium and oxygen (i.e. Ti/O ratio) estimated by integrating the area beneath the decomposed peaks of O(1s) (530.14 eV) and Ti(2p3/2) (458.74 eV) is ∼0.51 (i.e. Ti:O = 1.02:2), which is very close to the stoichiometric ratio (i.e. 1:2) of pure TiO2. This analysis confirms that all 1D nanoneedles in the large area array were fully oxidized, and were composed of pure stoichiometric TiO2 without any traces of sub-oxides (TiOx). Moreover, the binding energy difference (ΔE) of 71.33 eV between the O(1s) and Ti(2p3/2) peaks is very close to that of 71.5 eV for TiO2, and significantly smaller than 73.4 eV for Ti2O3 and 75.0 eV for TiO.28 This confirms again that the 1D nanoneedle array over a large area is formed exclusively of stoichiometric TiO2.
 | ||
| Fig. 2 Typical high-resolution XPS spectra of the (a) Ti(2p) and (b) O(1s) core levels of the large area array of 1D β-TiO2 nanoneedles. The XPS spectra were decomposed via Voigt curve function fittings. | ||
The supercapacitor behavior of 1D β-TiO2 nanoneedles is confirmed with CV, galvanostatic charge/discharge and impedance measurements. The CV measurements useful to investigate supercapacitor behavior are recorded for 1D β-TiO2 nanoneedles within the operating potential range of 0 to 0.8 V and at various scan rates ranging from 15 to 150 mV s−1. Fig. 3(a) shows the CV graphs of the 1D β-TiO2 nanoneedles. Irrespective of variation in the scan rates, the CV graph shows well rectangular shape in 1 M Na2SO4 electrolyte, indicating good capacitive behavior and high rate capability of the β-TiO2 nanoneedles. No evidence of any faradic reaction on well rectangular CV curve is found to ensure the reduction of Ti4+ to Ti3+. This confirms the pure double layer capacitor behavior of the β-TiO2 nanoneedles. Moreover, the well rectangular shape of CV graph confirms that the large surface area of the nanoneedle morphology of β-TiO2 with clearly visible textural boundaries offers abundant diffusion of Na+ ions and charge transport during non-faradic reaction along the nanoneedle sides. The negative and positive current density occurred due to the insertion and extraction of Na+ ions on the surface of β-TiO2 nanoneedles. A little bit more negative current density is observed than that of positive current density at applied potential. It is accepted that the large surface area of nanostructure morphology acts as traps to capture free electrons or ions when they pass through. Likewise, the large surface area of 1D TiO2 nanoneedles possibly provides a number of trap levels. Therefore, trapping–detrapping of free electrons at trap levels enhances the ionic conductivity17 and results in a more negative current density at applied potential. The current density of CV increased with increase in scanning rate from 15 to 150 mV s−1. Likewise, the area under CV graph increased proportionally with the scan rate. The variation in current density can be well understood via the Randles–Sevcik equation-
| ip = 2.69 × 105n3/2Coν1/2D1/2 | (2) |
 | ||
| Fig. 3 (a) Steady-state cyclic voltammograms of the 1D β-TiO2 nanoneedles obtained in the voltage range of 0 to 0.8 V at various scan rates from 15 to 150 mV s−1 in 1 M Na2SO4 electrolyte. (b) The graphical representation of the peak current density (ip) of the anodic reduction with the square root of the scan rate (ν). (c) The specific capacitance calculated from those cyclic voltammograms for various scan rates. | ||
The specific capacitance is shown in the Fig. 3(c) is calculated from CV graph at various scan rates (Fig. 3(a)) from the equation,
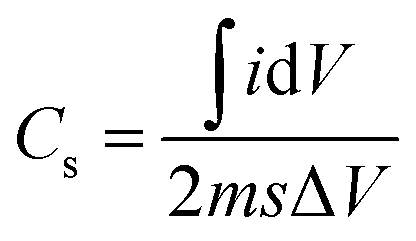 | (3) |
 is an integrated area of the CV curve. The calculated specific capacitance (Cs) varied linearly with the scan rate. The Cs value of 34.1 mF g−1 gained at the scan rate of 15 mV s−1 was retained to 31.6 mF g−1 after the scan rate of 150 mV s−1. The Cs retention of 92.7% was observed even after large variation in the scan rate. The capacitance of 34.1 mF g−1 might have resulted from maximum diffusion of Na+ ions over the large surface area provided by 1D nanoneedle array. The Cs value of 34.1 mF g−1 gained from 1D β-TiO2 is substantially larger than reported for TiO2 nanostructures and thin films such as amorphous and air annealed TiO2 nanotube arrays,7 polycrystalline anatase TiO2 nanowires,30 anatase/rutile mixed phase TiO2 nanocrystals,31 and rutile TiO2 nanorods.9 Although Salari et al.32 have reported enhancement of the Cs in anatase to rutile transformed TiO2 nanotubes via controlling oxygen vacancies, the very slow scan rate of 1 mV s−1 was necessary to achieve these values. Furthermore, 7.3% capacitance reduction of 1D β-TiO2 nanoneedles observed with an increased scan rate from 15 to 150 mV s−1, is a much smaller loss than the significant reduction reported in anodically oxidized self-organized TiO2 nanotubes (80.8% at 100 mV s−1),27 TiO2 nanowires and nanocrystalline powder (40.9 and 81.8% at 100 mV s−1, respectively),31 and seed-assisted hydrothermally grown rutile TiO2 nanowires (65% at 100 mV s−1).33 Moreover, TiO2 thin film (∼23.7% at 100 mV s−1) showed a reduction of ∼35.8% after preparing a multilayer film with graphene.34 The reason for this smaller reduction (i.e. 7.3%) in Cs of 1D β-TiO2 is instant diffusion/transport of ion along the textural boundaries of 1D β-TiO2 nanoneedles irrespective of scanning rate. Therefore, it is assumed that nanosize dimensions and increased textural boundaries of 1D β-TiO2 nanoneedles play a significant role in the diffusion of ion leading to the observed improvement.
is an integrated area of the CV curve. The calculated specific capacitance (Cs) varied linearly with the scan rate. The Cs value of 34.1 mF g−1 gained at the scan rate of 15 mV s−1 was retained to 31.6 mF g−1 after the scan rate of 150 mV s−1. The Cs retention of 92.7% was observed even after large variation in the scan rate. The capacitance of 34.1 mF g−1 might have resulted from maximum diffusion of Na+ ions over the large surface area provided by 1D nanoneedle array. The Cs value of 34.1 mF g−1 gained from 1D β-TiO2 is substantially larger than reported for TiO2 nanostructures and thin films such as amorphous and air annealed TiO2 nanotube arrays,7 polycrystalline anatase TiO2 nanowires,30 anatase/rutile mixed phase TiO2 nanocrystals,31 and rutile TiO2 nanorods.9 Although Salari et al.32 have reported enhancement of the Cs in anatase to rutile transformed TiO2 nanotubes via controlling oxygen vacancies, the very slow scan rate of 1 mV s−1 was necessary to achieve these values. Furthermore, 7.3% capacitance reduction of 1D β-TiO2 nanoneedles observed with an increased scan rate from 15 to 150 mV s−1, is a much smaller loss than the significant reduction reported in anodically oxidized self-organized TiO2 nanotubes (80.8% at 100 mV s−1),27 TiO2 nanowires and nanocrystalline powder (40.9 and 81.8% at 100 mV s−1, respectively),31 and seed-assisted hydrothermally grown rutile TiO2 nanowires (65% at 100 mV s−1).33 Moreover, TiO2 thin film (∼23.7% at 100 mV s−1) showed a reduction of ∼35.8% after preparing a multilayer film with graphene.34 The reason for this smaller reduction (i.e. 7.3%) in Cs of 1D β-TiO2 is instant diffusion/transport of ion along the textural boundaries of 1D β-TiO2 nanoneedles irrespective of scanning rate. Therefore, it is assumed that nanosize dimensions and increased textural boundaries of 1D β-TiO2 nanoneedles play a significant role in the diffusion of ion leading to the observed improvement.
The galvanostatic charging–discharging (Fig. S1, ESI†) of 1D β-TiO2 nanoneedles was studied at various current densities of 166.7, 250, 333.3 and 416.7 μA g−1. Obviously, the charging curves were relatively symmetric to their discharge counterpart implying that a highly reversible ion transportation is efficiently taking place along the textural boundaries of 1D β-TiO2 nanoneedles. Furthermore, the overall performance of 1D β-TiO2 nanoneedles was illustrated with a Ragone plot (ESI, Fig. S2†). A Ragone plot manifests a high energy density and power density of 3.04 W h kg−1, and 1683 W kg−1, respectively.
Not only the amount of active material and electrolyte plays a critical role in the capacitive behavior, but a dominant role is attributed to the electrical properties, textural boundaries, and the core of the nanostructures. Therefore, electrochemical impedance spectroscopy (EIS) was employed to investigate the mechanistic aspects such as electrical resistance involved in the ion diffusion of 1D β-TiO2 nanoneedle arrays. Typical Nyquist plots of the 1D β-TiO2 nanoneedle in 1 M Na2SO4 solutions at various electrode potentials are presented in Fig. 4. All the impedance spectra consist of partial semicircles (arc) and straight lines having slopes at the higher and lower frequencies, respectively. In high-frequency region, the distorted semicircle corresponds to the charge-transfer resistance of the interface between the 1D β-TiO2 nanoneedles and electrolyte. The intercept of this semicircle yields the electrolyte resistance (Re), and the diameter provides the charge transfer resistance (Rct). As noticed from the diameter of the semi-circle, the value of the Rct increased with the applied potential which reduces Cs. However, the low-frequency regime in the form of the straight line corresponds to diffusion resistance from the textural boundaries. Extrapolation of this gives larger resistance compare to that of the core of the nanoneedle since the 1D nanoneedle morphology of β-TiO2 provides large surface area. These observations are within the expectation and are supporting the calculated values of Cs. Moreover, the straight line observed in low-frequency regime represents Warburg behavior. The angle made by the low-frequency data on the real axis decreases gradually from 67.4° to 50.03° on increasing the applied potential from 0.2 to 0.8 V. This indicates gradual transition from capacitance to Warburg behavior between 0.2 and 0.8 V. The Warburg behavior (Warburg resistance Zw) results from the diffusion-controlled insertion and extractions of anions/cations in the electrode. This trend is in good agreement with the reports based on rutile TiO2 nanowires and TiO2@C core–shell nanowires,35 and anatase TiO2 nanotubes.8 The equivalent circuit for these electrochemical impedance measurements is composed of electrolyte resistance (Re), a charge transfer resistance (Rct), finite length Warburg diffusion element (Zw), and a constant phase element (CPE). It is represented in the inset of Fig. 4. Thus, nanoneedle morphology with large surface area and clearly visible textural boundaries improve the accessibility of the Na+ ions from the electrolyte. The time constant (τ) is calculated from the equation-
 | (4) |
 | ||
| Fig. 4 Electrochemical impedance spectra (Nyquist plot) of the 1D β-TiO2 nanoneedles acquired at various electrode potentials from 0 to 0.8 V. Inset shows best fitted equivalent circuit model for the impedance spectra of the 1D β-TiO2 nanoneedles. | ||
Good cycling stability, durability and lifetime are important characteristics for the highly stable performance of supercapacitors. The 1D β-TiO2 nanoneedles were tested at a scan rate of 100 mV s−1 and the current density of 250 μA g−1 for 10000 cycles and 5000 cycles, respectively (Fig. 5). The Cs evaluated from the CV measurements at a scan rate of 100 mV s−1 drops from 32.1 to 28.3 mF g−1 after 10000 cycles. Significantly, the 1D β-TiO2 nanoneedles exhibit long-term stability with only 11.8% reduction of Cs after 10000 continuous cycles (Fig. 5(a)). The CV graphs of selected cycles (Fig. S3†) used to evaluate the Cs retention are almost identical, indicating extremely stable performances of the 1D β-TiO2 nanoneedles. Furthermore, Cs measurements were performed for 5000 continuous cycles of galvanostatic charging–discharging at current density of 250 μA cm−2 in 200000 s (55.6 h) (Fig. 5(b)). Galvanostatic charging–discharging cycles were relatively symmetric to their discharge counterpart. First, 50 cycles are shown in Fig. S4.† With increased number of charging–discharging cycles and time, the calculated Cs drops from 109.9 to 96.7 mF g−1 after 5000 cycles. This small relative decrease indicates that 1D β-TiO2 nanoneedles exhibit long-term stability with only 12.04% reduction of Cs after continuous galvanostatic charging–discharging for 55.6 h (i.e. 5000 cycles). This ∼12% reduction of Cs of 1D β-TiO2 nanoneedles observed is much better than the value for amorphous TiO2 nanotubes (34.8% reduction after 10000 cycles) which were further annealed at 400 °C to form anatase TiO2 nanotubes (44.3% reduction after 10000 cycles),7 hydrothermally synthesized rutile TiO2 nanorods (20% reduction even after only 1000 cycles),9 sonochemically carbon activated TiO2 (β) nanowires mixed with polyaniline (57.5% reduction after 2000 cycles),4 polythiophene infiltrated TiO2 nanotubes (11% reduction after 1100 cycles),3 and arrays of anodic TiO2 nanotubes layered with Co(OH)2 by cathodic deposition (18% reduction after 1000 cycles).8 Moreover, these nanoneedles showed comparatively better stability than other metal oxides listed in ESI; Table S1.† Even after such a large number of cycles continued continuously for couples of days, 1D β-TiO2 nanoneedles were still intact and did not appear degraded or damaged by electrochemical interactions. This remarkable cycling performance of 1D β-TiO2 nanoneedles confirms their superior durability, the longer lifetime, high stability, and excellent electrochemical reversibility in Na2SO4 electrolyte. This is ascribed to their nanostructure, large surface area, clearly visible textural boundaries, enhanced electrical conductivity, and well stable electrochemical reactions. Therefore, the core–shell formation of 1D β-TiO2 nanoneedles with carbon based materials or conducting polymers may be helpful to overcome their swelling and shrinking and to further improve their stability at large extent.
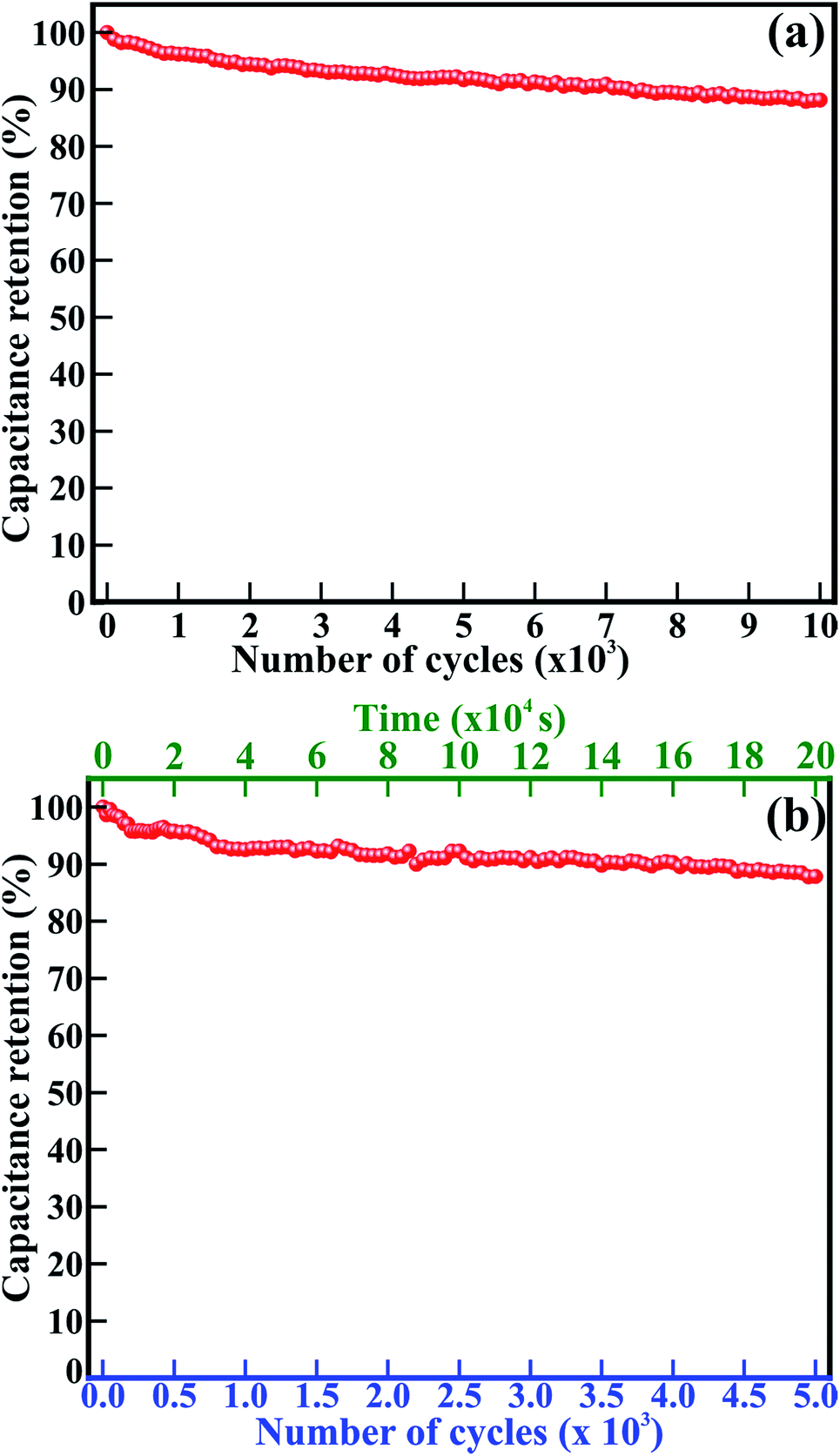 | ||
| Fig. 5 Cycling stability and capacitance retention of the 1D β-TiO2 nanoneedles observed by (a) cyclic voltammograms at a scan rate of 100 mV s−1 for 10000 cycles, and (b) galvanostatic charging–discharging at the current density of 250 μA g−1 for 5000 cycles of 55.6 h. The percentage numbers denote the specific capacitance retention with the increase in a number of cycles. | ||
Conclusions
Transparent large area array of 1D β-TiO2 nanoneedles were synthesized on conducting glass substrates via hot-filament metal vapor deposition. The 1D β-TiO2 nanoneedles of the orthorhombic crystalline structure of brookite were conical in shape and tapered in the downward direction with a diameter of about 24–26 nm and a length of ∼650 nm. The XPS double-peak features of the Ti(2p3/2) and Ti(2p1/2) core levels revealed that the large area array consisted exclusively of pure stoichiometric TiO2 nanoneedles (i.e. Ti:O = 1.02:2). The large area array of 1D β-TiO2 nanoneedles with large surface area and clearly visible textural boundaries provides distinct advantages of highly active surface sites and significantly facilitate for Na+ ion diffusion and charge transfer. The excellent electrochemical performance of large area arrays of 1D β-TiO2 nanoneedle in the electrolyte of Na+ ions delivered specific capacitance of 192.2 mF g−1 and 34.1 mF g−1 from CC and CV, respectively, high columbic efficiency (98%), energy density of 3.04 W h kg−1, and power density of 1683 W kg−1. The EIS tests confirmed the important role of textural boundaries of the 1D β-TiO2 nanoneedles in the enhancement of Na+ ion diffusion for highly stable non-faradic capacitance behavior. Additionally, the excellent cycle stability up to 10000 cycles makes the 1D β-TiO2 nanoneedle arrays an excellent material for the fabrication of supercapacitors with highly stable performance. Overall, the dimension and size-dependent electrochemical properties described in this work have opened up a new era in nanotechnology for the fabrication of supercapacitors with long cycle lifetime.
Acknowledgements
The authors would like to thank the Department of Science and Technology (DST), Ministry of Science and Technology of India, for their financial support of this research under INSPIRE Faculty award No. DST/INSPIRE Faculty Award/2013/IFA13-PH-63.References
- Z. S. Wu, K. Parvez, X. L. Feng and K. Mullen, Nat. Commun., 2013, 4, 2487 Search PubMed.
- L. H. Bao, J. F. Zang and X. D. Li, Nano Lett., 2011, 11, 1215–1220 CrossRef CAS PubMed.
- R. B. Ambade, S. B. Ambade, N. K. Shrestha, Y. C. Nah, S. H. Han, W. Lee and S. H. Lee, Chem. Commun., 2013, 49, 2308–2310 RSC.
- Q. Q. Tan, Y. X. Xu, J. Yang, L. L. Qiu, Y. Chen and X. X. Chen, Electrochim. Acta, 2013, 88, 526–529 CrossRef CAS.
- R. S. Devan, R. A. Patil, J. H. Lin and Y. R. Ma, Adv. Funct. Mater., 2012, 22, 3326–3370 CrossRef CAS.
- T. Brezesinski, J. Wang, J. Polleux, B. Dunn and S. H. Tolbert, J. Am. Chem. Soc., 2009, 131, 1802–1809 CrossRef CAS PubMed.
- X. H. Lu, G. M. Wang, T. Zhai, M. H. Yu, J. Y. Gan, Y. X. Tong and Y. Li, Nano Lett., 2012, 12, 1690–1696 CrossRef CAS PubMed.
- G. G. Zhang, C. J. Huang, L. M. Zhou, L. Ye, W. F. Li and H. T. Huang, Nanoscale, 2011, 3, 4174–4181 RSC.
- A. Ramadoss and S. J. Kim, J. Alloys Compd., 2013, 561, 262–267 CrossRef CAS.
- Q. Wang, Z. H. Wen and J. H. Li, Adv. Funct. Mater., 2006, 16, 2141–2146 CrossRef CAS.
- H. Zhou and Y. R. Zhang, J. Power Sources, 2013, 239, 128–131 CrossRef CAS.
- T. A. Kandiel, A. Feldhoff, L. Robben, R. Dillert and D. W. Bahnemann, Chem. Mater., 2010, 22, 2050–2060 CrossRef CAS.
- M. Posternak, A. Baldereschi, E. J. Walter and H. Krakauer, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 125113 CrossRef.
- S. D. Mo and W. Y. Ching, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 13023–13032 CrossRef CAS.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CAS.
- J. Z. Chen, W. Y. Ko, Y. C. Yen, P. H. Chen and K. J. Lin, ACS Nano, 2012, 6, 6633–6639 CrossRef CAS PubMed.
- H. Zhou and Y. Zhang, J. Phys. Chem. C, 2014, 118, 5626–5636 CAS.
- M. H. Yang, P. C. Chen, M. C. Tsai, T. T. Chen, I. C. Chang, H. T. Chiu and C. Y. Lee, CrystEngComm, 2014, 16, 441–447 RSC.
- H. Kominami, M. Kohno and Y. Kera, J. Mater. Chem., 2000, 10, 1151–1156 RSC.
- R. A. Patil, C. P. Chang, R. S. Devan, Y. Liou and Y. R. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 9872–9880 CAS.
- R. A. Patil, R. S. Devan, J. H. Lin, Y. Liou and Y. R. Ma, Sci. Rep., 2013, 3, 3070 Search PubMed.
- R. S. Devan, J. H. Lin, Y. J. Huang, C. C. Yang, S. Y. Wu, Y. Liou and Y. R. Ma, Nanoscale, 2011, 3, 4339–4345 RSC.
- R. S. Devan, C. L. Lin, S. Y. Gao, C. L. Cheng, Y. Liou and Y. R. Ma, Phys. Chem. Chem. Phys., 2011, 13, 13441–13446 RSC.
- R. S. Devan, J. H. Lin, W. D. Ho, S. Y. Wu, Y. Liou and Y. R. Ma, J. Appl. Crystallogr., 2010, 43, 1062–1067 CrossRef CAS.
- R. A. Patil, R. S. Devan, Y. Liou and Y. R. Ma, Sol. Energy Mater. Sol. Cells, 2016, 147, 240–245 CrossRef CAS.
- Y. Wang, H. J. Sun, S. J. Tan, H. Feng, Z. W. Cheng, J. Zhao, A. D. Zhao, B. Wang, Y. Luo, J. L. Yang and J. G. Hou, Nat. Commun., 2013, 4, 2214 Search PubMed.
- M. Salari, S. H. Aboutalebi, A. T. Chidembo, I. P. Nevirkovets, K. Konstantinov and H. K. Liu, Phys. Chem. Chem. Phys., 2012, 14, 4770–4779 RSC.
- W. B. Hu, L. P. Li, G. S. Li, C. L. Tang and L. Sun, Cryst. Growth Des., 2009, 9, 3676–3682 CAS.
- A. M. Zaky, S. S. A. El-Rehim and B. M. Mohamed, Int. J. Electrochem. Sci., 2006, 1, 17–31 CAS.
- J. Lee, J. Choi, J. Lee, S. K. Choi and H. D. Chun, Nanotechnology, 2005, 16, 1449–1453 CrossRef CAS.
- M. Salari, S. H. Aboutalebi, K. Konstantinov and H. K. Liu, Phys. Chem. Chem. Phys., 2011, 13, 5038–5041 RSC.
- M. Salari, K. Konstantinov and H. K. Liu, J. Mater. Chem., 2011, 21, 5128–5133 RSC.
- M. H. Yu, Y. X. Zeng, C. Zhang, X. H. Lu, C. H. Zeng, C. Z. Yao, Y. Y. Yang and Y. X. Tong, Nanoscale, 2013, 5, 10806–10810 RSC.
- W. W. Liu, X. B. Yan and Q. J. Xue, J. Mater. Chem. C, 2013, 1, 1413–1422 RSC.
- H. M. Zheng, T. Zhai, M. H. Yu, S. L. Xie, C. L. Liang, W. X. Zhao, S. C. I. Wang, Z. S. Zhang and X. H. Lu, J. Mater. Chem. C, 2013, 1, 225–229 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra11348f |
| This journal is © The Royal Society of Chemistry 2016 |