
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thiaporphyrin-mediated photocatalysis using red light†
JinGyu
Lee‡
,
James W.
Papatzimas‡
,
Ashley D.
Bromby
,
Evgueni
Gorobets
and
Darren J.
Derksen
*
Department of Chemistry, University of Calgary, 2500 University Drive NW, Calgary, Alberta T2N 1N4, Canada. E-mail: dderksen@ucalgary.ca
First published on 21st June 2016
Abstract
Penetration of light through biological tissue improves in the near-IR windows beyond 650 nm. In an effort to explore the chemical reactivity accessible using this low-energy radiation, this study reveals catalysts and reactions that are experimentally viable with red light. Thiophene-containing porphyrin compounds are capable of catalytic, photo-reductive dehalogenation on an array of α-halo ketone model substrates with low catalyst loadings (0.1 mol%), in the presence of low energy, red light (>645 nm).
Photo-mediated reactions have reemerged in synthetic chemistry as powerful tools to form and break chemical bonds.1 With major advances in the field, most light-mediated synthetic chemistry utilizes high energy blue or green light (375–570 nm). Recent developments in proximity-directed chemistry and photochemical release or activation of bioactive molecules, have validated the approach of using light to probe and interact with biological systems.2,3 The most commonly used photocatalysts in the field are ruthenium and iridium based polypyridyl complexes which absorb photons in the blue region of the visible spectrum (Fig. 1).4–6 Organic dyes such as eosin Y7 and rose bengal, which absorb in the green region, have also been reported in literature to be used as photocatalysts and show significant promise for synthetic applications.8,9 After identification of the synthetically tunable, thiophene-containing porphyrin scaffold, we decided to explore the photoreductive properties of meso-5,10,15,20-tetraphenyl-21-monothiaporphyrin (S1TPP), and 5,10,15,20-tetraphenyl-21,23-dithiaporphyrin (S2TPP) due to their absorbance properties at wavelengths beyond 650 nm and moderate excited state reduction potentials (−0.586 to −0.741 eV).10–13 Recent reports have also shown that S2TPP-related compounds, together with oxygen, have potential as light-activatable drug delivery systems using ‘photo-unclick’ chemistry.14 Based on our interest in exploring low-energy light reactivity within biological microenvironments, it was necessary to complete a model study on the reductive, bond-cleaving reactions that can occur using thiophene-containing porphyrins.
 | ||
| Fig. 1 Absorption properties of commonly used photocatalysts.8 | ||
We selected dehalogenation as a model system for exploring the use of red light for metal-free, photoreductive catalysis (Table 1). It was observed that alone, Ru(bpy)3Cl2 had very low reactivity under long wavelength light (entry 1). Combining Hantzsch ester (HEH) and Ru(bpy)3Cl2 provided a modest increase in yield (entry 2) above the reactivity of HEH without catalyst (entry 3).15 The addition of N,N-diisopropylethylamine (DIPEA) ensured consistent pH levels in the optimization reactions. Omission of catalyst and HEH resulted in a very low level of background dehalogenation (entry 4). In the absence of catalyst and base, the background reactivity of HEH was observed (entry 5). We immediately observed improved dehalogenation reactivity for S1TPP and S2TPP beyond reactions using Ru(bpy)3Cl2 at 0.1 mol% loading (entry 6,7). DIPEA plays a role in effective photocatalysis as reactions with only catalyst and HEH resulted in a decrease in yield (entry 8, 9). In combination with HEH, and base, yields of thiophene-mediated red light dehalogenation improved dramatically (entry 10, 11). Alternative bases triethyl- and tributyl amine were less effective than DIPEA (entry 12, 13) while the biomimetic NADH analog, 1-benzyl-1,4-dihydronicotinamide (BNAH), was the only alternate hydrogen atom source that provided effective reactivity – formic acid was ineffective in the reaction conditions (entry 14,15). To explore the possibility of a radical chain reaction-type mechanism, the reaction yield for bromoacetophenone under standard conditions was determined after 5 minutes of red-light irradiation (25% yield) that remained unchanged after an additional 17 hours in the dark (data not shown). Using the same model system, the addition of 5 eq. of TEMPO resulted in consumption of the Hantzsch ester and complete recovery of bromoacetophenone after 1 h. These results are not consistent with a radical chain reaction but instead support a typical photo-catalytic mechanism mediated by a hydrogen atom transfer agent1e (Scheme 1). Note that the dehalogenation model reaction of bromoacetophenone is complete within 1 h (73%) under the standard reaction conditions and is unaffected by the addition of 1 equivalent of water (74%). With the intent of reaching a balance between practicality and product yield, we selected a standard reaction time of 18 h to ensure low levels of substrate reactivity were not overlooked. Building on these preliminary results, we investigated the substrate scope on an array of α-functionalized carbonyl-containing compounds (Table 2). The influence of steric hindrance on dehalogenation efficiency for the thiophene-containing catalysts was evident based on decreasing isolated yields due to lower conversion and reduced reaction rates (entries 1–6). This analysis also revealed a trend that when S2TPP was used as the catalyst, isolated yields were consistently lower than S1TPP (40–60% vs. 75–90% respectively). These differences in reactivity are attributed to the stronger reduction potential,10 and marginally better solubility properties, of S1TPP. With reliably better yields, S1TPP was selected for further evaluation. Other halogens were also evaluated for reactivity under the optimized red-light conditions (entry 7, 8). Although chloroacetophenone showed some reactivity in the red-light optimized conditions, the rate of reaction and corresponding conversion are considerably lower than for bromoacetophenone. Similarly, the more labile iodoacetophenone reacted quantitatively, clearly demonstrating the effect of substrate bond-strength on the catalytic reactivity. No reactivity was observed on analogous ester or amide-derived substrates (entry 9, 10). Notably, under the standard reaction conditions, alkyl halo-ketones (entry 11, 12), also displayed modest conversion to the corresponding methyl ketones. There appears to be no inherent correlation between electronic effects of substituents as the overall yield is comparable for electron donating or withdrawing substituents (entry 13, 14). Aryl-bromides and nitriles remain compatible with the reaction conditions (entry 15, 16). The position of aryl functional groups and the effect of extended conjugation on reactivity were also explored (entry 17, 18). No by-products were detected during the course of our investigations, highlighting the advantage of using low energy photons for chemoselective bond cleavage. We have shown that thiophene-containing porphyrin catalysts are capable of directly catalyzing a modest scope of photo-reductive dehalogenation reactions using low energy red light above 645 nm. In contrast to many other photocatalytic dehalogenation reactions, this method does not require expensive transition metal catalysts for these dehalogenations to occur. Given the success of red-light mediated methods emerging in the recent literature, the further exploration of thiophene-based porphyrins for chemoselective light-mediated reactivity is warranted. We have optimized a reaction methodology that is capable of photochemically altering chemical bonds using light beyond 645 nm, deep into the red wavelengths of light. Further investigations toward expanding the substrate scope, and exploring chemoselectivity of the modified thiophene-containing porphyrins are currently underway.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Base | H-Atom source | Yield [%]b |
| a Standard conditions: 0.1 mol% catalyst, 1.1 eq. Hantzsch ester (HEH), 2 eq. DIPEA, DMF, 18 h, rt. b Yield determined through analysis with GC-MS. | ||||
| 1 | Ru(bpy)3Cl2 | DIPEA | None | 3 |
| 2 | Ru(bpy)3Cl2a | DIPEA | HEH | 18 |
| 3 | No catalyst | DIPEA | HEH | 13 |
| 4 | No catalyst | DIPEA | None | 3 |
| 5 | No catalyst | None | HEH | 21 |
| 6 | S1TPP | DIPEA | None | 26 |
| 7 | S2TPP | DIPEA | None | 25 |
| 8 | S1TPP | None | HEH | 13 |
| 9 | S2TPP | None | HEH | 10 |
| 10 | S1TPPa | DIPEA | HEH | 75 |
| 11 | S2TPPa | DIPEA | HEH | 72 |
| 12 | S1TPP | Et3N | HEH | 45 |
| 13 | S1TPP | Bu3N | HEH | 45 |
| 14 | S1TPP | DIPEA | HCO2H | 2 |
| 15 | S1TPP | DIPEA | BNAH | 58 |

 | ||
| Scheme 1 Proposed mechanism adapted from MacMillan and coworkers.1e HEH = Hantzsch ester, SET = single electron transfer. | ||
|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Substrate | Product | % yield | Entry | Substrate | Product | % yield | Entry | Substrate | Product | % yield |
| a Yield of purified product on 1 mmol scale.16 b S2TPP used as catalyst. c Yield determined through GC-MS analysis due to volatility of product. NR = no reaction, adam = adamantyl, biphenyl = 1-(1,1′-biphenyl)-4-yl. | |||||||||||
| 1 |
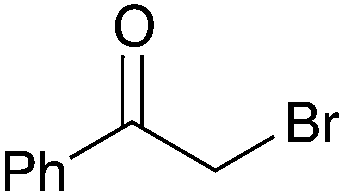
|
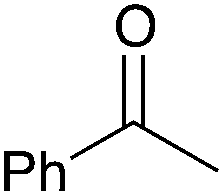
|
90 | 7 |

|

|
12 | 13 |

|
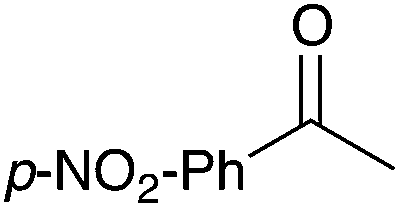
|
86 |
| 2 |

|
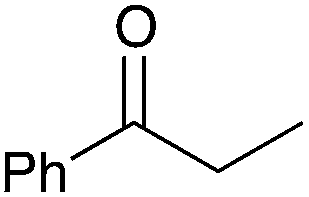
|
78 | 8 |

|

|
Quant | 14 |
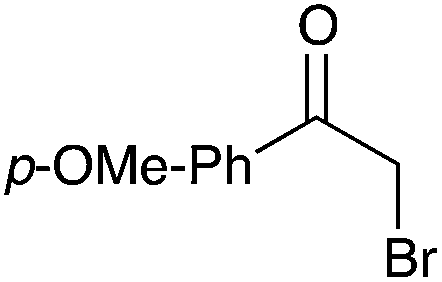
|

|
85 |
| 3 |

|

|
75 | 9 |
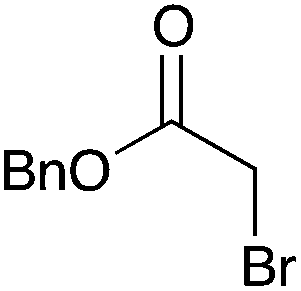
|
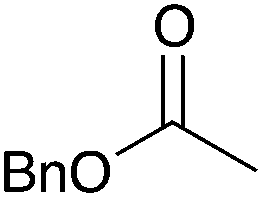
|
NR | 15 |
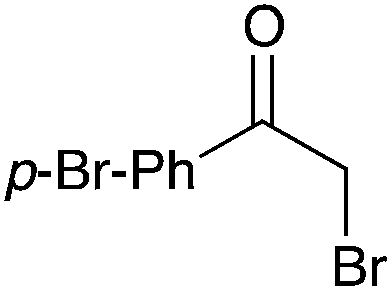
|

|
76 |
| 4b |

|

|
60 | 10 |

|

|
NR | 16 |
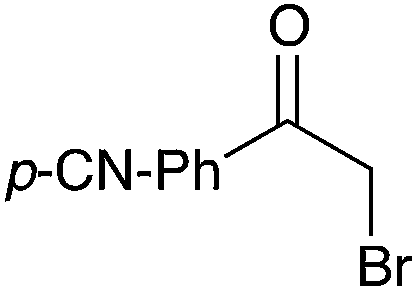
|

|
80 |
| 5b |
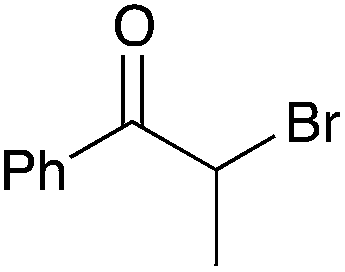
|

|
60 | 11 |

|
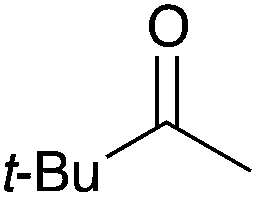
|
16c | 17 |

|
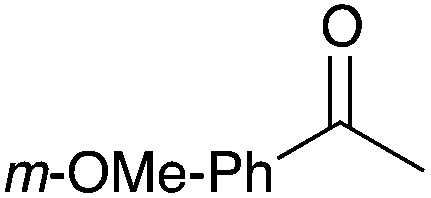
|
92 |
| 6b |
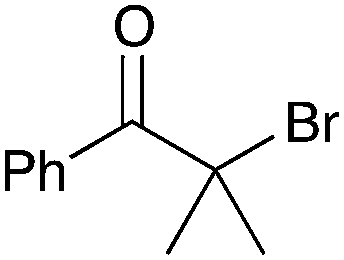
|

|
40 | 12 |

|

|
40 | 18 |

|
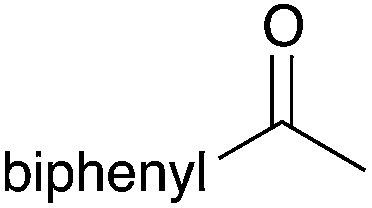
|
98 |

Acknowledgements
The authors wish to thank the Alberta Children's Hospital Research Institute, the Alberta Children's Hospital Foundation, the Charbonneau Cancer Institute and University of Calgary for their support of this research. Thanks to Mr Wade White and Mr Jian Jun Li in the University of Calgary, Department of Chemistry Instrumentation Lab. The authors would also like to thank Dr Todd Sutherland and Dr Belinda Heyne for helpful discussions in the development of this work.Notes and references
- For examples, see: (a) C. C. Nawrat, C. R. Jamison, Y. Slutskyy, D. W. C. MacMillan and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 11270 CrossRef CAS PubMed; (b) J. J. Devery and C. R. J. Stephenson, Nature, 2015, 519, 42 CrossRef CAS PubMed; (c) T. Herlig, T. Slanina, A. Hancock, U. Wille and B. König, Chem. Commun., 2015, 51, 6568 RSC; (d) L. R. Espelt, I. S. McPherson, E. M. Wiensch and T. P. Yoon, J. Am. Chem. Soc., 2015, 137, 2452 CrossRef PubMed; (e) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (f) J. C. T. Leung, C. Chatlova-Sazepin, J. G. West, M. Rueda-Becerril, J.-F. Paquin and G. M. Sammis, Angew. Chem., Int. Ed., 2012, 51, 10804 CrossRef CAS PubMed; (g) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (h) M. A. Ischay, J. Du and T. P. Yoon, Nat. Chem., 2010, 2, 527 CrossRef PubMed; (i) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785 CrossRef CAS PubMed; (j) M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886 CrossRef CAS PubMed; (k) S. Fukuzumi, S. Mochizuki and T. Tanaka, J. Phys. Chem., 1990, 94, 722 CrossRef CAS; (l) C. Pac, M. Ihama, M. Yasuda, Y. Miyauchi and H. Sakurai, J. Am. Chem. Soc., 1981, 103, 6495 CrossRef CAS.
- M. J. C. Long, J. R. Poganik and Y. Aye, J. Am. Chem. Soc., 2016, 138, 3610 CrossRef CAS PubMed.
- C. Gasser, S. Taiber, C.-M. Yeh, C. H. Wittig, P. Hegemann, S. Ryu, F. Wunder and A. Moglich, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8803 CrossRef CAS PubMed.
- For excitation wavelengths of a variety of transition metal photocatalysts, see: (a) L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143 CrossRef CAS; (b) M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712 CrossRef CAS; (c) J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J. Wang, S. Parker, R. Rohl, S. Bernhard and G. G. Malliaras, J. Am. Chem. Soc., 2004, 126, 2763 CrossRef CAS PubMed; (d) J. M. Kern and J. P. Sauvage, J. Chem. Soc., Chem. Commun., 1987, 546 RSC; (e) M. A. Haga, E. S. Dodsworth, G. Eryavec, P. Seymour and A. B. P. Lever, Inorg. Chem., 1985, 24, 1901 CrossRef CAS; (f) D. P. Rillema, G. Allen, T. J. Meyer and D. Conrad, Inorg. Chem., 1983, 22, 1617 CrossRef CAS; (g) R. J. Crutchley and A. B. P. Lever, J. Am. Chem. Soc., 1980, 102, 7128 CrossRef CAS; (h) R. C. Young, T. J. Meyer and D. G. Whitten, J. Am. Chem. Soc., 1976, 98, 286 CrossRef CAS.
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zewlesky, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS.
- D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688 RSC.
- For a tutorial on the redox properties of excited states and kinetics see: S. P. Pitre, C. D. McTiernan and J. C. Scaiano, Acc. Chem. Res., 2016 DOI:10.1021/acs.accounts.6b00012 For excitation wavelengths of a variety of organic photocatalysts, see: (a) I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725 CrossRef CAS PubMed; (b) W. Fu, W. Guo, G. Zou and C. Xu, J. Fluorine Chem., 2012, 140, 88 CrossRef CAS; (c) M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951 CrossRef CAS PubMed.
- K. Fidaly, C. Ceballos, A. Falguières, M. S.-I. Veitia, A. Guy and C. Ferroud, Green Chem., 2012, 14, 1293 RSC.
- R. P. Pandian, T. K. Chandrashekar, G. S. S. Saini and A. L. Verma, J. Chem. Soc., Faraday Trans., 1993, 89, 677 RSC.
- T. Kaur, W. Z. Lee and M. Ravikanth, Eur. J. Org. Chem., 2014, 11, 2261 CrossRef.
- A. Srinivasan, B. Sridevi, M. V. Ram Reddy, S. J. Narayanan and T. K. Chandrashekar, Tetrahedron Lett., 1997, 38, 4149 CrossRef CAS.
- D. G. Hilmey, M. Abe, M. I. Nelen, C. E. Stilts, G. A. Baker, S. N. Baker, F. V. Bright, S. R. Davies, S. O. Gollnick, A. R. Oseroff, S. L. Gibson, R. Hilf and M. R. Detty, J. Med. Chem., 2002, 45, 449 CrossRef CAS PubMed.
- (a) P. Thapa, M. Li, M. Bio, P. Rajaputra, G. Nkepang, Y. Sun, S. Woo and Y. You, J. Med. Chem., 2016, 59, 3204 CrossRef CAS PubMed; (b) M. Bio, P. Rajaputra, G. Nkepang, S. G. Awuah, A. M. L. Hossion and Y. You, J. Med. Chem., 2013, 56, 3936 CrossRef CAS PubMed.
- M. Neumann, S. Füldner, B. König and K. Zietler, Angew. Chem., Int. Ed., 2011, 50, 951 CrossRef CAS PubMed.
- To a solution of α-haloketone (1 mmol) in DMF (5 mL), HEH (279 mg, 1.1 equiv.), DIPEA (0.35 mL, 2 equiv.), and S1TPP, S2TPP, or Ru(bpy)3Cl2 (0.1 mol%) were added under stirring. The reaction vessel was kept in the dark until irradiation with red light. After 18 h of irradiation, the reaction mixture was diluted with water (8 mL) and extracted with Et2O (3 × 8 mL). The combined organic layer was washed with brine (20 mL), dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by silica gel flash column chromatography to give pure product. All reactions reported at rt remained at a temperature of 22 °C ± 2 °C over the course of the reaction.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra11374e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |