
Functionalization of pyridyl ketones using deprotolithiation-in situ zincation†
Madani Hedidi‡
ab,
William Erb*a,
Frédéric Lassagnea,
Yury S. Halauko*c,
Oleg A. Ivashkevichc,
Vadim E. Matulisd,
Thierry Roisnele,
Ghenia Bentabed-Ababsa*b and
Florence Mongina
aChimie et Photonique Moléculaires, Institut des Sciences Chimiques de Rennes, UMR 6226, Université de Rennes 1-CNRS, Bâtiment 10A, Case 1003, Campus de Beaulieu, 35042 Rennes, France. E-mail: william.erb@univ-rennes1.fr
bLaboratoire de Synthèse Organique Appliquée, Faculté des Sciences, Université d'Oran 1 Ahmed Ben Bella, BP 1524 El M'Naouer, 31000 Oran, Algeria. E-mail: badri_sofi@yahoo.fr
cUNESCO Chair of Belarusian State University, 14 Leningradskaya Str., Minsk 220030, Belarus. E-mail: hys@tut.by
dResearch Institute for Physico-Chemical Problems of Belarusian State University, 14 Leningradskaya Str., Minsk 220030, Belarus
eCentre de Diffractométrie X, Institut des Sciences Chimiques de Rennes, UMR 6226, Université de Rennes 1-CNRS, Bâtiment 10B, Campus de Beaulieu, 35042 Rennes, France
First published on 28th June 2016
Abstract
The metallation of aryl ketones was achieved by using LiTMP in the presence of ZnCl2·TMEDA, as evidenced by subsequent interception with iodine or by a palladium-catalysed cross-coupling reaction. One of the synthesized iodo ketones has been further elaborated to reach derivatives of biological interest.
Pyridines play a large part in biological processes (nicotine, niacin, NADP, vitamin B6…), in pharmaceuticals and agrochemicals,1 as well as in organic materials.2 In addition, pyridyl ketones bearing a halogen at the position adjacent to the carbonyl function are key intermediates to access heterocyclic scaffolds of interest such as azafluorenones,3 azaxanthones,4 naphthyridones,5 and thieno-,6 pyrazolo-7 and isoxazolo-pyridines.8
Even if deprotonative lithiation9 has been largely used to regioselectively functionalize pyridines,10 the method has never been extended to pyridyl ketones due to their low compatibility with organolithiums. Mixed lithium–nonalkali metal combinations have been developed to achieve chemoselective deprotometallation of aromatics.11 In this context, the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of homometallic amides LiTMP (TMP = 2,2,6,6-tetramethylpiperidino) and Zn(TMP)2,12 obtained by mixing in a 3:1 ratio LiTMP and ZnCl2·TMEDA (TMEDA = N,N,N′,N′-tetramethylethylenediamine) in THF (THF = tetrahydrofuran), was successfully employed with a large range of sensitive substrates.13 Such a synergy, attributed to reversible deprotolithiation shifted by zinc-mediated transmetallation12 (or ‘trans-metal trapping’14), has since been extended to the use of LiTMP in the presence of ZnCl2·2LiCl, MgCl2 or CuCN·2LiCl.15
:1 mixture of homometallic amides LiTMP (TMP = 2,2,6,6-tetramethylpiperidino) and Zn(TMP)2,12 obtained by mixing in a 3:1 ratio LiTMP and ZnCl2·TMEDA (TMEDA = N,N,N′,N′-tetramethylethylenediamine) in THF (THF = tetrahydrofuran), was successfully employed with a large range of sensitive substrates.13 Such a synergy, attributed to reversible deprotolithiation shifted by zinc-mediated transmetallation12 (or ‘trans-metal trapping’14), has since been extended to the use of LiTMP in the presence of ZnCl2·2LiCl, MgCl2 or CuCN·2LiCl.15
Herein, we report the efficiency of aryl ketones as directing groups for LiTMP-mediated deprotometallation of pyridines and other arenes in the presence of ZnCl2·TMEDA as in situ trap (Scheme 1, Table 1).
 | ||
| Scheme 1 Zincation of aryl ketones 1 using LiTMP in the presence of ZnCl2·TMEDA followed by iodolysis or palladium-catalysed cross-coupling to afford ketones 2 and 2′m. | ||
| Entry | Substrate/n/temperature | Product/yielda (%) |
|---|---|---|
| a Yield after purification by column chromatography.b 4-Benzoyl-3,5-diiodopyridine (2b′) was also obtained in 10% yield.c 1,5-Diiodo-4-azafluorenone (2j′) was also obtained in 20% yield.d 1,6-Diiodo-4-azaxanthone (2k′) was also obtained in 10% yield. | ||
| 1 |  |
 |
| 2 |  |
 |
| 3 |  |
 |
| 4 |  |
 |
| 5 | 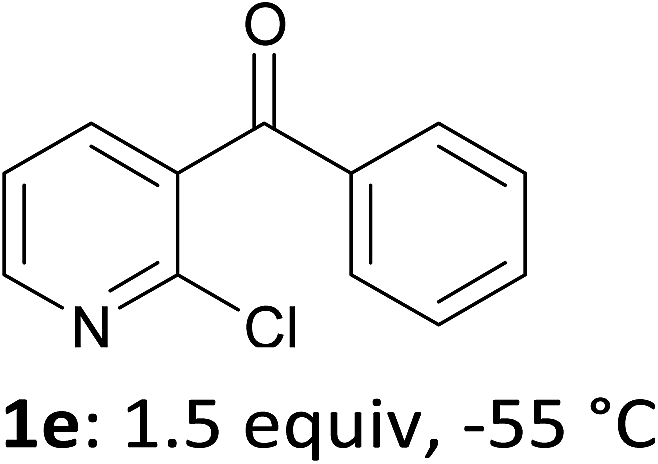 |
 |
| 6 | 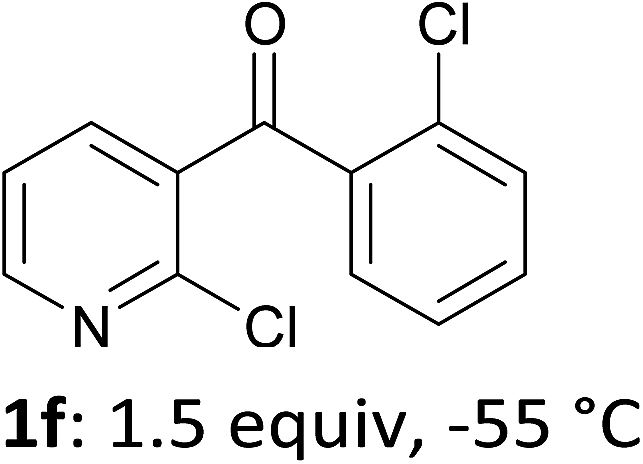 |
 |
| 7 |  |
 |
| 8 | 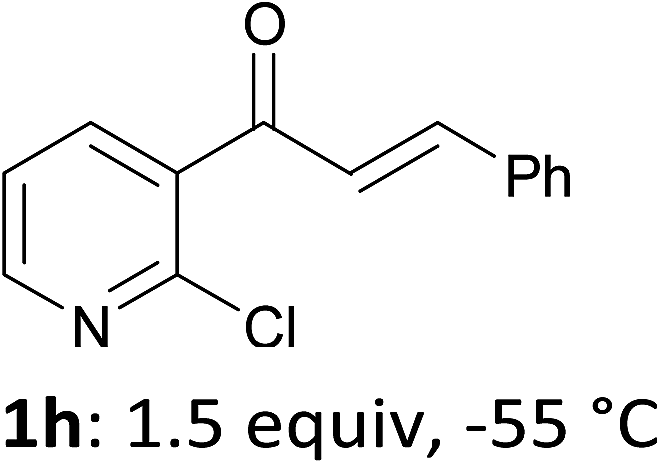 |
 |
| 9 |  |
 |
| 10 |  |
 |
| 11 |  |
 |
| 12 |  |
 |
Thus, after optimization of the reaction conditions (using four different reaction temperatures from −70 to −10 °C and different amounts of LiTMP from 1 to 3 equiv.), treatment of 2-benzoylpyridine (1a) in THF containing ZnCl2·TMEDA (1 equiv.) with LiTMP (1.5 equiv.) at −30 °C for 15 min and then iodine led to the 3-iodo derivative 2a in 50% yield (entry 1). Similarly, 4-benzoylpyridine (1b) was converted to the 3-iodo derivative 2b by using LiTMP (2 equiv.); with this substrate benefiting from two free positions adjacent to the benzoyl group, a second deprotonation was observed to some extent, as evidenced by the competitive formation of the diiodo 2b′ (entry 2).
3-Benzoylpyridine (1c) is more prone to nucleophilic attack onto the ring than its 2- and 4-isomers.16 As a consequence, deprotolithiation-zincation could only be evidenced by subsequent iodolysis at temperatures below −50 °C. Using LiTMP (1.5 equiv.) at −55 or −70 °C for 15 min and then iodine provided the 4-iodo derivative 2c in 30 or 37% yield, respectively (entry 3).
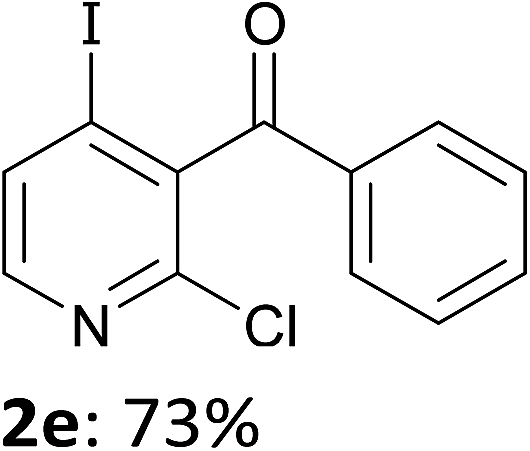
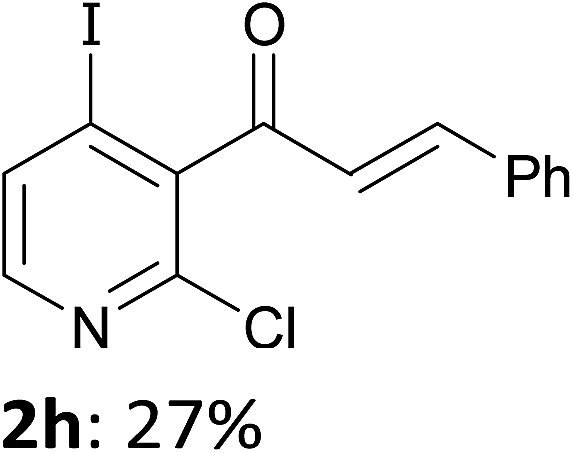
When present at pyridine 2-position, halogens are known to acidify the 4-position, as shown by pKa values calculated in THF.13a The 2-halogeno 3-benzoylpyridines 1d–h were thus prepared.17 Accordingly, when similarly reacted by using LiTMP at −55 °C, the iodo derivatives 2d–g were isolated in improved yields, in line with the long range effects of fluorine and chlorine (entries 4–7). In contrast, the reaction from 1h proved more complex, giving the iodide 2h in a modest yield (entry 8).
The presence of a methoxy group at 2-position of 3-benzoylpyridine also had a positive impact on the course of the reaction since involving 1i in the sequence furnished the iodo derivative 2i in 88% yield (entry 9). Whereas this group does not acidify remote pyridine 4-position,13b it acts by making the pyridine ring less sensitive towards competitive nucleophilic attack. Indeed, based on the 1H NMR chemical shifts in CDCl3 of 1c (8.12 and 8.82 ppm for H4 and H6, respectively) and 1i (7.72 and 8.32 ppm for H4 and H6, respectively), one can predict as shown by Handy and Zhang18 that the partial positive charges at C4 and C6 will be reduced for 1i.
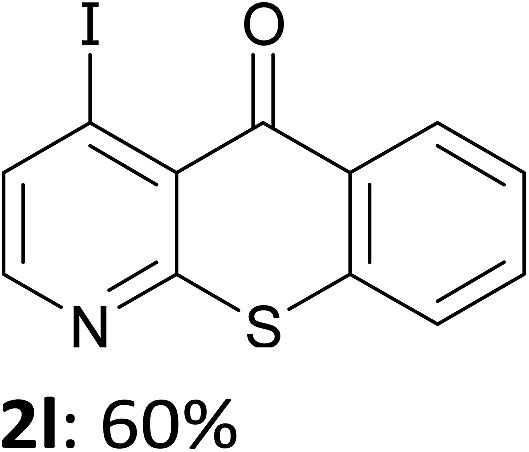
The behaviour of the ketones 1j–l, possessing reduced flexibility on carbonyl direction, was similarly examined. 4-Azafluorenone (1j) was converted to the 1-iodo derivative 2j in a moderate yield, similar to that obtained from 3-benzoylpyridine (1c). In the case of 1j, the phenyl ring was also attacked to a lesser extent at a position facing the pyridine nitrogen to afford the diiodide 2j′ (entry 10, Fig. 1). In contrast with the reaction from 1j, the sequence using 4-azaxanthone (1k) and 4-azathioxanthone (1l) provided the iodides 2k and 2l in higher 60% yields (entries 11 and 12).
 | ||
| Fig. 1 ORTEP diagrams (30% probability) of compounds 2j′ and 2k′. | ||
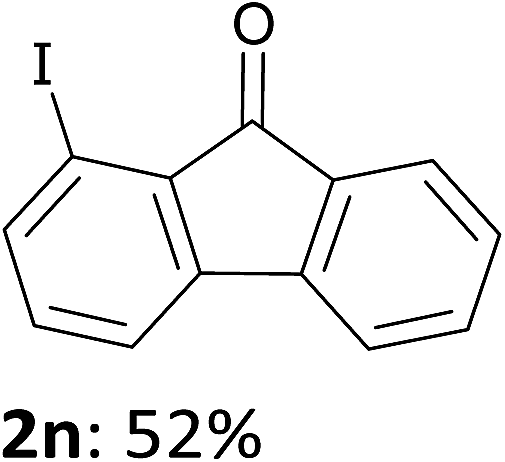
In order to evaluate the scope of the method, we chose other aromatic substrates.19 When compared with its 4-aza analogue 1k, xanthone (1m) similarly led to the 1-iodo 2m in 72% yield. On the contrary, as previously noted between 1j and 1k, the reaction from 1n was less efficient than from 1m. Indeed, 1-iodofluorenone (2n) was obtained in 52% yield, but together with the ketone 2n′ (35% yield) resulting from an addition of the metallated product to 1n (Scheme 1, Table 2). As organozincs hardly react with ketones, the product 2n′ could rather result from an addition of 1-lithiofluorenone to 1n more rapid than its trapping by the zinc species. These results suggest that the carbonyl direction in the metallated derivative coming from 1n is less prone to stabilize a 1-lithio compound than that from 1m.





Interestingly, the deprotolithiation-in situ zincation could be combined with a subsequent Negishi cross-coupling reaction.20 Thus, using catalytic amounts of PdCl2 as palladium source and 1,1′-diphenylphosphinoferrocene (dppf) as ligand with 2-chloropyridine12a allowed 2′m to be formed from 9-xanthone (1m) in 76% yield (Scheme 1).
CH acidities are in general useful data to better understand deprotometallation outcomes, in particular regarding regioselectivity issues. We thus calculated selected pKa values in THF solution by means of quantum chemistry at the DFT B3LYP level of theory.13 After geometry optimization and calculation of the vibrational frequencies by using the 6-31G(d) basis set, the single point energies were obtained using the 6-311+G(d,p) basis set. The solvent influence was treated through the polarized continuum model (PCM) with the default parameters for THF. Finally, the pKa values were reached from the Gibbs energy of the homodesmic reaction between the studied and probe heterocycles.
That both sets of CH acidity values for 1m and 1n are rather similar (Table 2) supports the role of the carbonyl direction on the course of the reaction through coordination. Analogously, the difference observed between the pKa values of 1j and 1k is not significant enough to allow for a rationalization of the distinct yields noted, rather suggesting a more efficient stabilization of the metallated compound by the carbonyl group in the case of 1k at the origin of the higher yield. Besides, the formation of dimetallated products from 1j and 1k, as demonstrated by isolation of 2j′ and 2k′ (Fig. 1), could be favoured for the former by a nitrogen assistance and, for the latter, by a relatively low pKa value (35.6) at the 6-position (Table 1).
Finally, when 2-benzoylthiophene (1o) was submitted to LiTMP in the presence of ZnCl2·TMEDA as before, the reaction took place next to sulphur to afford after iodolysis the iodo derivative 2o in 80% yield (Scheme 2).
 | ||
| Scheme 2 Zincation of phenyl 2-thienyl ketone (1o) using LiTMP in the presence of ZnCl2·TMEDA followed by iodolysis to afford the iodinated 2-thienyl ketone 2o. | ||
To move towards nitrogen-containing derivatives of biological interest, 2-benzoyl-3-iodopyridine (2a) was involved in further reactions (Scheme 3). A catalyst–base system was first optimized to perform guanidine copper-catalysed N-arylation.21 Upon treatment with K3PO4 as base and CuI as catalyst source in the presence of DMSO, the iodide 2a was converted to the pyrido[3,2-d]pyrimidine 3a in 77% yield. Besides, cyclizing Suzuki coupling22 was performed from the ketone 2a and 2-aminophenylboronic acid under palladium catalysis to provide 5-phenylbenzo[f][1,7]naphthyridine (4a)23 in 68% yield.
 | ||
| Scheme 3 Conversion of 2-benzoyl-3-iodopyridine (2a) to derivatives of biological interest. | ||
In summary, we have reported a short and simple access to iodinated aryl ketones. Additionally, transition metal-mediated reactions occurring with cyclization led to elaborated scaffolds. Applications towards the synthesis of libraries of compounds for biological evaluation are currently under investigation in our laboratory.
Acknowledgements
We thank the Ministère de l'Enseignement supérieur et de la Recherche scientifique Algérien (M. H.), the Centre national de la recherche scientifique and the Institut Universitaire de France (F. M.). We acknowledge FEDER founds (D8 VENTURE Bruker AXS diffractometer) and Thermo Fisher (generous gift of 2,2,6,6-tetramethylpiperidine). This research has been partly performed as part of the CNRS PICS project “Bimetallic synergy for the functionalization of heteroaromatics”.Notes and references
- T. Eicher, S. Hauptmann and A. Speicher, The Chemistry of Heterocycles, Wiley-VCH, New York, 2nd edn, 2003 Search PubMed.
- See for example: (a) M. Kim and J. Y. Lee, Chem.–Asian J., 2012, 7, 899 CrossRef PubMed; (b) H.-W. Lin, C.-W. Lu, L.-Y. Lin, Y.-H. Chen, W.-C. Lin, K.-T. Wong and F. Lin, J. Mater. Chem. A, 2013, 1, 1770 RSC.
- (a) N. Marquise, V. Dorcet, F. Chevallier and F. Mongin, Org. Biomol. Chem., 2014, 12, 8138 RSC; (b) D.-W. Gao, C. Zheng, Q. Gu and S.-L. You, Organometallics, 2015, 34, 4618 CrossRef CAS.
- (a) F. Marsais, F. Trécourt, P. Bréant and G. Queguiner, J. Heterocycl. Chem., 1988, 25, 81 CrossRef; (b) G. A. Eller, V. Wimmer, A. W. Haring and W. Holzer, Synthesis, 2006, 4219 CrossRef CAS.
- (a) M. Y. Platts, C. G. Barber, J.-Y. Chiva, R. L. Eastwood, D. R. Fenwick, K. A. Paradowski and D. C. Blakemore, Tetrahedron Lett., 2011, 52, 512 CrossRef; (b) S. Massari, B. Mercorelli, L. Sancineto, S. Sabatini, V. Cecchetti, G. Gribaudo, G. Palu, C. Pannecouque, A. Loregian and O. Tabarrini, ChemMedChem, 2013, 8, 1403 CrossRef CAS PubMed.
- (a) K. Kobayashi, T. Kozuki and H. Konishi, Heterocycles, 2009, 78, 2993 CrossRef CAS; (b) K. Kobayashi, T. Suzuki and Y. Egara, Helv. Chim. Acta, 2013, 96, 69 CrossRef CAS.
- (a) E. Bisagni, M. Rautureau and C. Huel, Heterocycles, 1989, 29, 1815 CrossRef CAS; (b) M. Atobe, K. Naganuma, M. Kawanishi, A. Morimoto, K.-i. Kasahara, S. Ohashi, H. Suzuki, T. Hayashi and S. Miyoshi, Bioorg. Med. Chem. Lett., 2014, 24, 1327 CrossRef CAS PubMed.
- E. J. Hanan, R. V. Fucini, M. J. Romanowski, R. A. Elling, W. Lew, H. E. Purkey, E. C. VanderPorten and W. Yang, Bioorg. Med. Chem. Lett., 2008, 18, 5186 CrossRef CAS PubMed.
- (a) H. W. Gschwend and H. R. Rodriguez, Org. React., 1979, 26, 1 CAS; (b) P. Beak and V. Snieckus, Acc. Chem. Res., 1982, 15, 306 CrossRef CAS; (c) V. Snieckus, Chem. Rev., 1990, 90, 879 CrossRef CAS; (d) T. G. Gant and A. I. Meyers, Tetrahedron, 1994, 50, 2297 CrossRef CAS; (e) M. Schlosser, Organometallics in Synthesis, ed. M. Schlosser, 2nd edn, 2002 Search PubMed.
- (a) G. Queguiner, F. Marsais, V. Snieckus and J. Epsztajn, Adv. Heterocycl. Chem., 1991, 52, 187 CrossRef CAS; (b) F. Mongin and G. Queguiner, Tetrahedron, 2001, 57, 4059 CrossRef CAS; (c) M. Schlosser and F. Mongin, Chem. Soc. Rev., 2007, 36, 1161 RSC.
- (a) R. E. Mulvey, Acc. Chem. Res., 2009, 42, 743 CrossRef CAS PubMed; (b) B. Haag, M. Mosrin, H. Ila, V. Malakhov and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9794 CrossRef CAS PubMed; (c) F. Mongin and A. Harrison-Marchand, Chem. Rev., 2013, 113, 7563 CrossRef CAS PubMed. A few carbonyl-containing five-membered heteroaromatics proved compatible: (d) W. Lin, O. Baron and P. Knochel, Org. Lett., 2006, 8, 5673 CrossRef CAS PubMed; (e) S. H. Wunderlich and P. Knochel, Angew. Chem., Int. Ed., 2007, 46, 7685 CrossRef CAS PubMed; (f) M. Mosrin and P. Knochel, Org. Lett., 2009, 11, 1837 CrossRef CAS PubMed; (g) T. Bresser, M. Mosrin, G. Monzon and P. Knochel, J. Org. Chem., 2010, 75, 4686 CrossRef CAS PubMed.
- (a) J. M. L'Helgoual'ch, A. Seggio, F. Chevallier, M. Yonehara, E. Jeanneau, M. Uchiyama and F. Mongin, J. Org. Chem., 2008, 73, 177 CrossRef PubMed; (b) P. García-Álvarez, R. E. Mulvey and J. A. Parkinson, Angew. Chem., Int. Ed., 2011, 50, 9668 CrossRef PubMed.
- For previous examples in the pyridine series, see: (a) K. Snégaroff, T. T. Nguyen, N. Marquise, Y. S. Halauko, P. J. Harford, T. Roisnel, V. E. Matulis, O. A. Ivashkevich, F. Chevallier, A. E. H. Wheatley, P. C. Gros and F. Mongin, Chem.–Eur. J., 2011, 17, 13284 CrossRef PubMed; (b) M. Hedidi, G. Bentabed-Ababsa, A. Derdour, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis, F. Chevallier, T. Roisnel, V. Dorcet and F. Mongin, Tetrahedron, 2016, 72, 2196 CrossRef CAS.
- M. A. Fuentes, A. R. Kennedy, R. E. Mulvey, J. A. Parkinson, T. Rantanen, S. D. Robertson and V. Snieckus, Chem.–Eur. J., 2015, 21, 14812 CrossRef CAS PubMed.
- M. R. Becker and P. Knochel, Angew. Chem., Int. Ed., 2015, 54, 12501 CrossRef CAS PubMed.
- For Lewis acid-catalysed nucleophilic additions at C4 onto 3-substituted pyridines, see: Q. Chen, X. Mollat du Jourdin and P. Knochel, J. Am. Chem. Soc., 2013, 135, 4958 CrossRef CAS PubMed.
- T. T. Nguyen, N. Marquise, F. Chevallier and F. Mongin, Chem.–Eur. J., 2011, 17, 10405 CrossRef CAS PubMed.
- S. T. Handy and Y. Zhang, Chem. Commun., 2006, 299 RSC.
- Concerning the deprotonation of benzophenone using a lithium–cadmium base, see: K. Snégaroff, J. M. L'Helgoual'ch, G. Bentabed-Ababsa, T. T. Nguyen, F. Chevallier, M. Yonehara, M. Uchiyama, A. Derdour and F. Mongin, Chem.–Eur. J., 2009, 15, 10280 CrossRef PubMed.
- On the topic, see: (a) E. Negishi, A. O. King and N. Okukado, J. Org. Chem., 1977, 42, 1821 CrossRef CAS; (b) E. Negishi, Acc. Chem. Res., 1982, 15, 340 CrossRef CAS.
- For reviews on the topic, see: (a) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954 CrossRef CAS PubMed; (b) I. P. Beletskaya and A. V. Cheprakov, Organometallics, 2012, 31, 7753 CrossRef CAS.
- For reviews on the topic, see: (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (b) S. Kotha, K. Lahiri and D. Kashinath, Tetrahedron, 2002, 58, 9633 CrossRef CAS.
- Y.-F. Chen and J.-C. Hsieh, Org. Lett., 2014, 16, 4642 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General procedures, experimental procedures and compound characterizations, 1H and 13C NMR spectra of the new compounds, and X-ray crystallographic data. CCDC 1475309 (2j′), 1475009 (2k′) and 1475010 (2n′). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra11370b |
| ‡ Present address: Département de Chimie, Faculté des Sciences, Université Hassiba Benbouali de Chlef, Hay Es-Salem, RN 19, 02000 Chlef, Algeria. |
| This journal is © The Royal Society of Chemistry 2016 |