DOI:
10.1039/C6RA11347H
(Paper)
RSC Adv., 2016,
6, 100732-100742
Amorphous nanosized Al–Ti–Mn trimetal hydrous oxides: synthesis, characterization and enhanced performance in arsenic removal†
Received
2nd May 2016
, Accepted 12th October 2016
First published on 13th October 2016
Abstract
Arsenite (As(III)) is more toxic and more difficult to remove from water than arsenate (As(V)). There is no simple treatment for direct efficient removal of As(III), and thus pre-oxidation of As(III) to As(V) is always required to achieve acceptable removal rates. However, this leads to a complicated operation, which is not cost-effective. To overcome these disadvantages, we have developed a novel nanosized Al–Ti–Mn trimetal hydrous oxide (ATM) adsorbent material combining the oxidation properties of manganese dioxide and the high capacity of aluminium hydrous oxide and titanium hydrous oxide to adsorb As(V). This was done by applying a method based on simultaneous oxidation and precipitation. The adsorbent was characterized by BET surface area measurement, as well as XRD, TEM, XPS, FTIR and TGA techniques. The characterization provided evidence that the new adsorbent was amorphous, had a relatively high surface area (71 m2 g−1) and consisted of aggregates of many nanosized particles. Laboratory experiments were carried out to investigate the adsorption kinetics, the adsorption capacity of the adsorbent and the effect of the pH of the solution on the overall arsenic removal. The results indicated that the ATM adsorbent performed excellently in removing both As(III) and As(V) from water. The maximum adsorption capacities for As(III) and As(V) calculated from the Langmuir model are 202.7 and 146.7 mg g−1 (pH 7.0), and 193.3 and 158.6 mg g−1 (pH 5.0), respectively. These values exceed significantly those reported in the literature for other nano-adsorbents. Furthermore, the result of XPS analysis of the adsorbent before and after interaction with As(III) confirmed the oxidation/adsorption mechanism for As(III) uptake by ATM. The comparison of –OH density (OH per g) in fresh ATM and ATM after adsorption of As(III) and As(V) shows that the number of –OH groups on the adsorbent surface was the key factor affecting its adsorption capacity. The amorphous nanosized Al–Ti–Mn trimetal hydrous oxide is thus a promising adsorbent for both As(III) and As(V) removal because of its excellent performance and simple low-cost synthesis process.
1. Introduction
Arsenic (As) pollution in drinking water and its side effects on human health have raised increasing concern globally over the past decade.1 More than 150 million people worldwide ingest excessive amounts of arsenic through drinking water contaminated from both natural geogenic and anthropogenic sources.2 Many highly populated Asian countries like Bangladesh, India, China and Vietnam are known to be affected by high groundwater arsenic concentrations.3 Unlike organic pollutants, arsenic does not decay, and thus circulates and eventually accumulates within the human body, which may result in a variety of health problems, including various forms of cancer (e.g. skin, lung, and bladder), cardiovascular and peripheral vascular diseases.4,5 The International Agency for Research on Cancer (IARC) has classified arsenic as a Class A human carcinogen. Controlled-ingestion studies in humans indicate that both As(III) and As(V) are well absorbed from the gastrointestinal tract.6,7 In view of the negative effects of arsenic on humans and the environment, the World Health Organization (WHO) has recommended a limit of 10 μg L−1 of arsenic for drinking water, which has been adopted by many nations as a regulatory standard.4 Arsenic mainly exists in four oxidation states (−III, 0, +III and +V), although the predominant forms of As in soil and water are arsenate (As(V)) and arsenite (As(III)).8 Depending on the redox state of the water environment, the valence states of arsenic vary from place to place. Arsenate, As(V), is more prevalent in oxygenated surface waters, while arsenite, As(III), is more likely to occur in anaerobic groundwater.9,10 As(III) is more toxic than As(V) and also more difficult to remove from aqueous environments.8,11–13 The common strategy for As(III) removal is based on its oxidation to As(V) by various techniques.8,14–16 Once oxidized to As(V), arsenic can be removed more easily, e.g. by adsorption.8,17
Therefore, developing an economical, effective and reliable treatment technique for arsenic removal from groundwater is critical and has gained considerable attention in recent years. Various treatment techniques such as coagulation/precipitation,18 hybrid process of ion exchange membrane-coagulation,19 adsorption9,20 and membrane processes19 have been developed and employed for arsenic removal. Due to its simplicity, high efficiency and cost-effectiveness, adsorption processes are regarded as the most promising methods and largely used for arsenic removal from water and wastewater.8,21 Different adsorbents, including natural and synthetic materials, have been extensively investigated to remove arsenic from aqueous solutions.8,22,23 Most recently, the development of composite adsorbents containing two or more metal oxides has gained considerable attention.24–27 Some of the recent results have demonstrated that mixed oxides are more active than single oxide thanks to the synergistic effects between the metals in heterogeneous mixed oxides.24–26 It is well known that aluminium, titanium and manganese hydroxides exist ubiquitously in natural aquatic systems and play an important role in geochemical cycling of nutrients and contaminants.28–30 Because of their low cost and environmental friendliness, these oxides are also widely used in water treatment engineering systems as important adsorbents for removing contaminants.15,31–33 Recently, it was found that the incorporation of aluminum, titanium or manganese into the adsorbents can significantly improve their adsorption capacity toward arsenic because they have a unique selectivity for polyoxy anions. For instance, Tina Basu et al.34 developed a synthetic bimetallic iron(III)–aluminum(III) oxide (NHIAO) adsorbent, which has a much higher As(V) adsorption capacity than the individual Al and Fe oxide adsorbents. Zhang et al.35 prepared Fe–Mn binary oxide adsorbent, exhibiting a significant enhancement in both As(V) and As(III) removal. Gupta and Ghosh17 reported the synthesis and use of an Fe–Ti binary mixed oxide for arsenic adsorption. Li et al.36 synthesized a Ce–Ti oxide adsorbent with high efficiency in both As(V) and As(III) removal. Kun Wu et al.37 found that an Mn oxide – doped Al oxide prepared in their laboratory had a high adsorption capacity towards arsenite and arsenate as compared to the results of recent published research. It is noteworthy that MnO2 component in the composite adsorbents FMBO, MNFHO and MODAO35,37,38 plays the role of an oxidant in the As(III) uptake through the following simplified reaction:
| | |
MnO2 + H3AsO3 + 2H+ → Mn2+ + H3AsO4 + H2O
| (1) |
In addition, the photocatalyzed oxidation of As(III) on TiO2 offers an environmentally benign method for arsenic removal.39 Thus the components significantly improved the performance of other pure metal oxides for As(III) removal. From the published results, it can be confirmed that the physico-chemical properties of multi-metal oxides differ from those of their single component oxides.40 It is also believed that the differences are the primary reason for the improved adsorption performance of multi-metal oxides and these materials may become promising adsorbents for arsenite oxidation and arsenate adsorption from contaminated waters in water treatment systems. To the best of our knowledge, although the characteristics of arsenic adsorption by single-component Al, Ti or Mn hydrous oxides have been studied and reported,41,42 there is little information available about the adsorption behavior of the composite of trimetal hydrous oxide. Therefore, the ATM adsorbent was recently developed by a simple chemical coprecipitation method in our laboratory and is reported in this study. The effect of various adsorbent characteristics, such as adsorption isotherms, adsorption kinetics and the effect of pH on arsenic adsorption, were investigated in batch experiments. The surface characteristics of this adsorbent were also studied by FTIR, XPS and TGA to elucidate the mechanisms for arsenite oxidation and arsenate adsorption.
2. Material and methods
2.1. Materials
All chemicals were analytical grade and were used without further purification (Sigma Aldrich). Reaction vessels (glass) were cleaned with 1% HNO3 and rinsed several times with deionized water before use. As(V) and As(III) stock solution was prepared by dissolving NaHAsO4·7H2O and NaAsO2 in distilled water, respectively. As(V) and As(III) working solutions were freshly prepared by diluting arsenic stock solutions with NaNO3 solution.
2.2. Synthesis of nanosized ATM
ATM was prepared using the oxidation and coprecipitation method. The main preparation procedure was briefed as follows: potassium permanganate (KMnO4, 0.95 g) was dissolved in 100 mL of deionized water; aluminum nitrate nonahydrate (Al(NO3)3·9H2O, 8.43 g), manganese sulfate monohydrate (MnSO4·xH2O, 1.53 g) and titanium oxysulfate (TiOSO4·xH2O, 7.19 g) were dissolved in another 200 mL of deionized water. Under vigorous magnetic stirring, the mixture solution of Al(NO3)3·9H2O, MnSO4·xH2O and TiOSO4·xH2O was added into the KMnO4 solution with simultaneous dropwise addition of 4 M NaOH to keep the solution pH in the range of 7.5–8. After addition, the suspension that formed was continuously stirred for 1 h, aged at room temperature for 12 h, and then washed several times with deionized water. The suspension was then filtered and dried at 105 °C for 24 h. The dry material was crushed and stored in a desiccator for further use. As a reference, a sample containing titanium and manganese was also prepared by a similar process.
2.3. Characterization of nanosized ATM
X-ray diffraction was performed using a Philips APD 1700, Almelo, using copper Kα radiation. The specific surface area of ATM was measured by nitrogen adsorption using the BET method with a Micromeritics ASAP 2000 (Micromeritics Co., USA) surface area analyzer. The particle size of the adsorbent was determined by a laser particle size analyser (Mastersizer 2000, Malvern Co., UK). Structural characterization was carried out using a Transmission Electron Microscope (JEOL JEM-1010, USA). The point of zero charge (PZC) was measured via a slightly modified method described by Kinniburgh et al.43 FTIR spectra were collected using a Thermo Scientific Nicolet iS10 FT-IR Spectrometer applying the ATR technique. The IR spectra of the original adsorbent and arsenic-loaded adsorbent were obtained as dry samples. All IR measurements were carried out at room temperature. The photoelectron spectra of the samples were measured using an ESCA3 MkII (VG Scientific, UK) electron spectrometer equipped with a hemispherical electron analyzer operated at constant pass energy of 20 eV and giving an energy resolution, expressed by the FWHM of Au 4f7/2 line, of 1.2 eV. The binding energy scale was calibrated using Au 4f7/2 (84.0 eV) and Cu 2p3/2 (932.6 eV) photoemission lines. The spectra were calibrated by setting C 1s peak belonging to adventitious carbon on the sample surface at the binding energy of 284.8 eV. Al Kα radiation was used for electron excitation. A small amount of powder sample was spread on a clean copper surface. The spectra were recorded at room temperature. The Al 2p, Mn 3p, Mn 2p, Ti 3p, Ti 2p, As 3d, O 1s and C 1s photoelectrons were measured. The electron detection angle was 45° with respect to the macroscopic sample surface. The pressure of residual gases in the analyser chamber during spectra acquisition was 2 × 10−9 mbar. The accuracy of the measured electron energies was ±0.2 eV. The overlapping spectral features were resolved into individual components using the damped non-linear least squares method44 after subtraction of Shirley background45 using Gaussian–Lorentzian line shape. Quantification of the elemental concentrations was accomplished by correcting photoelectron peak intensities for their cross-sections.46 In calculations homogeneous composition of the analyzed layer of the measured sample was assumed. Thermal gravimetric study was performed using a 10 mg sample in an alumina pan by a TG-DTA (Discovery DSC, TA Instruments, Delaware, USA).
2.4. Batch adsorption
Adsorption isotherms, adsorption kinetics and the effect of solution pH on arsenic adsorption were determined in batch adsorption experiments. For adsorption isotherms, the experiments were performed at pH 5.0 and 7.0. The pH of the suspensions was adjusted with 0.1 M of NaOH and/or HNO3 during the experiment. Initial arsenic concentration varied from 10 mg L−1 to 500 mg L−1. In each test, 30 mg of ATM were loaded in a 100 mL glass vessel, to which 50 mL of a solution containing differing amounts of arsenic were then added. The vessels were shaken on an orbit shaker at 300 rpm for 24 h at 25 ± 1 °C. Then, all samples were filtered by a 0.45 μm membrane filter and analyzed for arsenic. For the adsorption kinetics experiments, defined amount of As(III) or As(V) stock solution was added in a 1000 mL glass vessel containing 500 mL 0.01 M NaNO3 solution to prepare the initial arsenic concentration of 20 mg L−1. The solution pH was adjusted to 7.0 ± 0.1 by adding 0.1 M HNO3 and/or NaOH and then ATM was added to obtain a 0.3 g L−1 suspension. The suspension was mixed by magnetic-stirring, and the pH was maintained at 7.0 ± 0.1 throughout the experiment by addition of the acid and base solutions. In the whole process, only several drops of acid or base were added into the solution and the total volume was no more than 0.5 mL, which did not significantly influence the arsenic adsorption. Approximately 20 mL aliquots were taken from the suspension at certain time intervals. The samples were filtered through a 0.45 μm membrane filter and analysed for arsenic.
To investigate the influence of pH on arsenic adsorption, experiments were carried out by adding 30 mg of ATM into 150 mL glass vessels containing 50 mL of 50 mg L−1 As(III) or As(V) solution. The pH of the solutions was adjusted every four hours with dilute HNO3 or NaOH solution to designated values during the adsorption process. The equilibrium pH was measured and the supernatant was filtered through a 0.45 μm membrane after the solutions have been mixed for 24 h. Then, the residual arsenic concentration in the supernatants was determined. To evaluate the adsorption mechanism of As(III) and As(V) by FTIR, XPS and TGA techniques, the two solid samples were evaluated by adding 90 mg of ATM into 250 mL glass vessels, containing 150 mL of 300 mg L−1 As(III) or As(V) solution. Having been shaken for 24 h, the suspension was then filtrated and dried. The dry samples were stored and denoted as ATM + As(III) and ATM + As(V).
2.5. Analytical methods
After filtration by a 0.45 μm membrane filter, the concentration of residual arsenic concentrations of all experiments was measured using an inductively coupled plasma atomic emission spectroscopy machine (Agilent 720/725 ICP-OES, Agilent co., Australia). Prior to analysis, the aqueous samples were stored in acid-washed glass vessels. All samples used in our analysis were analysed within 12 h after collection.
3. Results and discussion
3.1. Characterization of ATM
The X-ray diffraction pattern of the adsorbent is illustrated in Fig. S1a.† The XRD spectrum of ATM showed no peaks, which suggested that the co-precipitation process inhibited the formation of any crystalline phase. The distribution of particle size of the powdered ATM is depicted in Fig. S1b,† indicating that the average particle size of the trimetal hydrous oxide is 20.76 μm. Fig. 1a and b illustrates the TEM image of the oxide particles, showing that they are aggregates formed by small nanoparticles (around 28–53 nm) with no sign of crystallinity. This indicates, in combination with XRD data, that ATM is totally X-ray amorphous. The value of point of zero charge (PZC) of ATM is approximately 3.45. The FTIR, TGA (see section 3.5) and XRD results showed that ATM is X-ray amorphous hydrous oxide of Al, Ti and Mn. The X-ray amorphous ATM has a relatively high BET surface area of 71 m2 g−1 and a pore volume of 0.329 cm3 g−1, in which micropore volume and mesopore volume are 0.0335 cm3 g−1 and 0.2093 cm3 g−1, respectively (Fig. S2 and S3†).
 |
| | Fig. 1 TEM images of ATM at different magnifications. | |
3.2. Adsorption kinetics
The pseudo-first order model and pseudo-second order model were employed to describe the kinetic data. The mathematical representations of the models are given in eqn (2) and (3), respectively.| |
 | (2) |
| |
 | (3) |
where qe and qt are the amount of adsorbed arsenic per unit weight of adsorbent (mg g−1) in equilibrium and at time t, respectively. k1, is the rate constant of pseudo-first order kinetic adsorption (min−1) as calculated from the gradient of the linear plot of log(qe − qt) versus t. k2 is the rate constant of pseudo-second order kinetic adsorption as calculated from the slope of the linear plot of t versus t/qt. In addition, the initial adsorption rate h can be obtained by the following equation:47
The gradient and intercept values were obtained by the least-squares regression method. The arsenic adsorption kinetic tests were conducted to determine the adsorption rate.
Fig. 2a shows the As(III) and As(V) removal kinetic profiles with an adsorbent loading of 0.3 g L−1 under neutral conditions. The adsorption of As(V) occurred more rapidly and required less time to reach equilibrium than that of As(III). The kinetics parameters were obtained through fitting the experimental data and are summarised in Table 1. According to the values of regression coefficients (R2), the pseudo-second order model fitted better, which indicates that the adsorption process might be chemisorption (Fig. 2b and Table 1). The initial adsorption rate (h) of As(V) and As(III) onto the ATM was 10.246 and 3.559 mg g−1 min−1, respectively (Table 1). The initial adsorption rate h (mg g−1 min−1) could be used as an indicator of the adsorption rate, especially at the beginning of the adsorption process. It is clear that the initial adsorption rate of As(V) on the adsorbent was higher than that of As(III), indicating that the ATM adsorbent removed As(V) faster than As(III). The relatively slow adsorption rate of As(III) may be caused by the oxidation step before the adsorption of As(V) as evidenced by the XPS results (see section 3.5). In addition, at pH = 7.0 ± 0.1, the dominant As(V) species are negatively charged (H2AsO4− and HAsO42−), when the dominant As(III) species is neutrally charged (H3AsO3). The negatively charged As(V) species are more easily removed than As(III).48,49 The adsorption behavior of As(V) and As(III) on the ATM was similar to that on the adsorbents reported previously, such as iron(III)–tin(IV) binary mixed oxides.50
 |
| | Fig. 2 (a) Adsorption kinetics of As(III) and As(V) on the ATM at pH = 7.0 ± 0.1. Arsenic initial concentrations = 20 mg L−1, adsorbent dose = 0.3 g L−1 and T = 25 ± 1 °C. (b) Pseudo-second order kinetic model fitting for the removals of As(III) and As(V) by the ATM. The fitted parameters are summarized in Table 1. | |
Table 1 Adsorption rate constants obtained from pseudo-first order model and pseudo-second order model
| Arsenic species |
Pseudo-first order model |
Pseudo-second order model |
| qexp (mg g−1) |
k1 (min−1) |
qe (mg g−1) |
R2 |
qe (mg g−1) |
h (mg g−1 min−1) |
R2 |
| As(III) |
50.10 |
0.00299 |
13.96 |
0.9579 |
50.00 |
3.559 |
0.9995 |
| As(V) |
49.70 |
0.00230 |
7.69 |
0.7324 |
49.75 |
10.246 |
0.9999 |
3.3. Adsorption isotherms
The arsenic adsorption capacities of the ATM at the two pH values were evaluated using the isotherms and are presented in Fig. 3. It can be seen that the ATM has high adsorption capacity for both As(III) and As(V). For As(V), its adsorption capacity was slightly reduced when the pH of the solution was increased from 5.0 to 7.0. In contrast, in the case of As(III) the increase in pH slightly enhanced its adsorption. In addition, at low initial equilibrium concentration, the adsorbent is more effective in removing As(V) than As(III). For example, the As(III) and As(V) adsorption capacities are 69.7 mg g−1 and 76.0 mg g−1 at initial concentration of 50 mg L−1 and pH 7.0. However, it has higher adsorption capacity for As(III) than that of As(V) at higher equilibrium concentration. These results indicate that the adsorption of As(III) and As(V) might be dominated by different mechanisms. Both Langmuir and Freundlich models were used to describe the adsorption isotherms. The Langmuir equation and Freundlich equation are represented as eqn (5) and (6), respectively.| |
 | (5) |
where qe is the amount of arsenic adsorbed on the solid phase (mg g−1), Ce is the equilibrium arsenic concentration in solution phase (mg L−1), KL is the equilibrium adsorption constant related to the affinity of binding sites (L mg−1), qm is the maximum amount of the arsenic per unit weight of adsorbent for complete monolayer coverage, KF is roughly an indicator of the adsorption capacity and n is the heterogeneity factor – the more heterogeneous the surface, the lower the value of the factor.
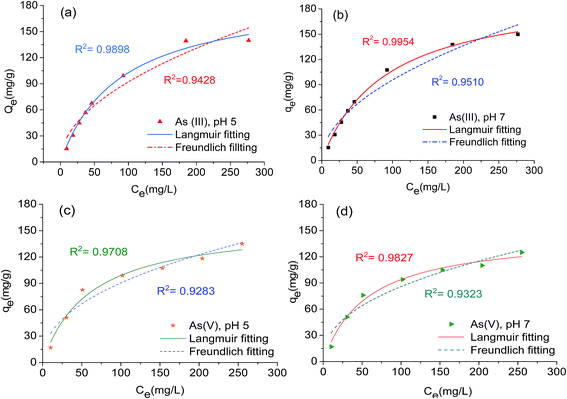 |
| | Fig. 3 Adsorption isotherms for (a and b) As(III) and (c and d) As(V) by ATM at pH 5 and pH 7 (T = 25 ± 1 °C; (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) Langmuir model fitting and ( ) Langmuir model fitting and (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) Freundlich model fitting). ) Freundlich model fitting). | |
The adsorption constants obtained from the isotherms are presented in Table 2. As shown there, the higher regression coefficient suggests that the Langmuir model is more suitable than the Freundlich model for describing how arsenic is adsorbed by ATM. The Langmuir model assumes that adsorption occurs on a homogeneous surface. Interestingly, although ATM consists of aluminum, titanium and manganese hydrous oxides, it exhibited the behavior typical for adsorption of a homogeneous surface having adsorption sites with similar adsorption energies. The maximal adsorption capacities for As(III) and As(V) calculated from Langmuir model are 202.7 and 146.7 mg g−1 (pH 7.0), and 193.3 and 158.6 mg g−1 (pH 5.0), respectively.
Table 2 Langmuir and Freundlich isotherm parameters for As(III) and As(V) adsorption on ATM at pH 5.0 and pH 7.0
| As species and pH |
Langmuir model |
Freundlich model |
| qm (mg g−1) |
KL (L mg−1) |
R2 |
KF (g L−1) |
n |
R2 |
| As(III), pH 5.0 |
193.28 |
0.0113 |
0.9898 |
9.00 |
0.5050 |
0.9428 |
| As(III), pH 7.0 |
202.72 |
0.0111 |
0.9954 |
9.05 |
0.5121 |
0.9510 |
| As(V), pH 5.0 |
158.58 |
0.0166 |
0.9708 |
12.00 |
0.4380 |
0.9283 |
| As(V), pH 7.0 |
146.72 |
0.0177 |
0.9827 |
12.14 |
0.4246 |
0.9323 |
To the best of our knowledge, these values are among the highest adsorption capacities for arsenic reported in literature (Table 3). Such high capacity indicates that the prepared ATM is very effective for both As(III) and As(V) removal.
Table 3 Comparison of maximum arsenic adsorption capacities for different adsorbents
| Adsorbent |
As con. range (mg L−1) |
Max. As(III) adsorption capacity (mg g−1) |
Max. As(V) adsorption capacity (mg g−1) |
Reference |
| MnO2 nanorods |
0–200 |
19.41 |
n.a. |
51 |
| Al2O3/Fe(OH)3 |
0.56–10.1 |
9.0 (pH 6.6) |
36.7 (pH 7.2) |
52 |
| Nano-TiO2 |
0–130 |
99.0 (pH 7.0) |
n.a. |
53 |
| CuO nanoparticles |
0–100 |
26.9 (pH 8.0) |
22.6 (pH 8.0) |
22 |
| Fe–Ti oxides |
5.0–250 |
85.0 (pH 7.0) |
14.3 (pH 7.0) |
54 |
| Fe–Mn binary oxide |
0–40 |
100.4 (pH 6.9) |
53.9 (pH 6.9) |
35 |
| Mn oxide doped Al oxide |
1–90 |
142.2 (pH 7.0) |
99.7 (pH 7.0) |
37 |
| Fe3O4:Cu |
1–85 |
37.97 (pH 5.0) |
42.90 (pH 5.0) |
55 |
| Fe–Zr trimetal oxide |
0–40 |
120.0 (pH 7.0) |
46.1 (pH 7.0) |
56 |
| Al–Ti–Mn trimetal hydrous oxide |
0–200 |
193.3 (pH 5.0) |
158.6 (pH 5.0) |
This study |
| Al–Ti–Mn trimetal hydrous oxide |
0–200 |
202.7 (pH 7.0) |
146.7 (pH 7.0) |
This study |
3.4. The influence of pH on arsenic adsorption
The results in Fig. 4 illustrate the effects of pH on the removal of As(V) and As(III). In case of As(V) adsorption, negligible variation in As(V) capacity was observed in the pH range of 3.3–6.1, while a further pH increase up to 10 resulted in an obvious drop in As(V) uptake. This phenomenon can be explained by electrostatic and ligand exchange mechanisms. The pH of the solution not only affected the surface charge of the ATM, but also influenced arsenic speciation in solution. In aqueous solutions, H2AsO4− and HAsO42− are the dominant As(V) species under the tested pH range (3–10). The adsorbent had a point of zero charge at pH 3.45, suggesting that the adsorbent surface was negative at pH above 3.45 and positive at pH below 3.45. Lower pH is favorable for the protonation of the adsorbent surface. Increased protonation is thought to enlarge the attraction force existing between the adsorbent surface and anionic arsenic species, and therefore to increase the amount of As(V) adsorbed in the lower pH region. In the higher pH region, the negatively charged sites dominate, the repulsion effect is stronger and the amount of adsorption consequently drops. On the other hand, as illustrated schematically by the reactions given in the eqn (7) and (8) below and by Le Chatelier's principle, when the pH increases, there are more hydroxyl ions (OH−) present in the solution, which increases the competition for adsorption sites; therefore, the surface adsorption of As(V) is lowered. On the contrary, the reduction of pH facilitates the ligand exchange by adsorption, resulting in increased arsenic removal. Similar phenomena were also observed for the adsorption of arsenic onto other Fe–Mn binary oxides26 and MnO2 nanorods.51 The reason is that the adsorption of strong acid anions by metal oxides and hydroxides typically decreases with an increasing pH.57
 |
| | Fig. 4 Effect of solution pH on As(III) and As(V) adsorption by ATM. The initial arsenic concentration = 50 mg L−1, adsorbent dose = 0.6 g L−1 and T = 25 ± 1 °C. | |
The same trend was observed with the As(III) species. Generally, As(III) adsorption capacity decreases with an increase in pH of the solution. The maximum adsorption of As(III) occurred under acidic conditions, remained relatively stable at the pH range of 5–8 and decreased markedly with further increase in pH. It was quite different from other adsorbents such as Fe–Zr binary oxide56 or ZrO2 nanoparticle.58 Such different pH dependence implies that As(III) adsorption on ATM adsorbent may be driven by a different mechanism. As illustrated later (Section 3.5), As(III) removal by ATM contains two basic processes, oxidation of As(III) to As(V) by Mn-oxides and adsorption of As(V) by metal oxides. As shown in eqn (1), low pH is favorable for As(III) oxidation into As(V) by Mn oxides, while high solution pH resulted in a smaller amount of As(III) oxidized into As(V). On the other hand, after the oxidation process, the increase of solution pH is also unfavorable for removal of As(V) species as described above by electrostatic and ligand exchange mechanism.
In addition, it is necessary to mention that although the adsorption capacity of ATM towards As(III) and As(V) was reduced gradually with the increase in pH, except for the pH range of 5–8, the adsorption capacities remained relatively high and stable, which is different from the other adsorbents reported previously 8, 20, 23, 51. This behavior has practical implications for determining the adsorbent's usability in the treatment of drinking water.
3.5. Influence of coexisting anions on adsorption
The effects of commonly present anions such as CO32−, SO42− and HPO42− in water on arsenic adsorption were examined at different concentrations (from 20 to 100 mg L−1). The results are shown in Fig. 5. The presence of HPO4− hinders significantly the adsorption of both As(III) and As(V); especially at high concentration levels this may be due to a strong competition for the binding sites of the adsorbent between HPO42− and arsenic. The presence of CO32− and HPO42− increased the pH of the solution (Fig. S7†), thus the high concentration of CO32− and HPO42− had a negative effect on As(V) removal since the ATM adsorbent exhibited the highest adsorption capacity at low pH. On contrary, the increase in CO32− concentration and the resulting higher pH of the solution facilitated the adsorption of As(III) (Fig. 5a and S7†). It can be hence suggested that the adsorption of As(III) is more sensitive to the change of pH solution than to the effect of coexisting CO32− anions. The presence of SO42− at different concentrations has only a minor effect on the pH of the solution and on the adsorption capacity.
 |
| | Fig. 5 Effects of co-existing anions on (a) As(III), (b) As(V) adsorption at different concentrations. Initial anion species concentration 50 mg L−1, concentration of co-existing anion 20–100 mg L−1, adsorbent dose 0.6 g L−1 and T = 25 ± 1 °C. | |
3.6. Evaluation of the adsorption mechanism by XPS, FTIR and TGA techniques
XPS and FTIR analyses were employed to explore the interactions between As(III), As(V) and the adsorbent surface, and the obtained results are shown in Fig. 6 and 7. XPS is a useful tool for the determination of concentration and chemical state (i.e. valence) of elements present in the surface layers of the studied samples.59,60 As shown in Fig. 6a, the wide scan XPS spectrum of the fresh adsorbent indicates presence of Al, Ti, Mn, C, O and small amounts of S on the adsorbent surface (originated from the precursor metal salt). New As 3d core level peak as well as As LMM and As 3p peaks appeared in the spectra of ATM after its interaction with As(V) or As(III), indicating clearly the presence of arsenic on the surface of the samples. The elemental concentrations (in atomic%) before and after adsorption calculated from XP spectra are shown in Table S1.† Fig. 6b illustrates the As 3d spectra of the ATM after adsorption of As(III) and As(V). The same binding energy of 45.5 eV is observed for the As 3d lines of arsenic loaded ATM regardless of the arsenic state present in the solution. The value is consistent with the values reported61,62 for arsenic in oxidation state (+V). Thus, it is inferred that As(III) species were completely oxidized to As(V) on the surface of the ATM. ATM played a role of catalyst for As(III) → As(V) reaction and the Mn-hydrous oxide component might function as the oxidant.9 As shown in Table S2,† the binding energies of electrons in Mn(II), Mn(III) and Mn(IV) oxidation states of manganese are close and the spectra of Mn 3p and Mn 2p photoelectrons are rather complex.63,64 Consequently, the population of individual oxidation states from the measured spectra cannot be unequivocally ascertained. The measured binding energy of Mn 3p electrons, 50.2 ± 0.2 eV is within the experimental error not influenced by adsorption of As and corresponds to (IV) oxidation state of manganese.65 The shift in the Mn 2p3/2 spectra of about 0.5 eV towards lower binding energy is observed for the ATM + As(III) sample (Fig. 6c). The lower kinetic energy of the Mn 2p3/2 electrons compared to Mn 3p electrons results in higher surface selectivity of the Mn 2p3/2 electrons and the observed shift thus indicate presence of Mn (+III)66,67 in the uppermost layer of the ATM + As(III) sample (for MnO2 and Mn2O3 the inelastic mean free path of Mn 3p electrons calculated from the TPP 2M formula68 is 2.7 nm, while for Mn 2p3/2 electrons it amounts to 1.8 nm). It was also reported that manganese oxides acted as a catalyst for the oxidation of organic compounds and inorganic ions.9,69 The spectra of O 1s photoelectrons (Fig. 6d) consist of two components, one belonging to oxygen in the ATM structure (at 530.2 eV) assigned to O2− and the other pertaining to hydroxyl groups (at 532.1 eV) assigned to OH− on the surface.70 The population of –OH groups decreased markedly after adsorption of As(III) and As(V). The details are listed in Table 4. This suggests that –OH groups on the adsorbent surface played an important role in the arsenic adsorption and were replaced by arsenic species.
 |
| | Fig. 6 XPS spectra of ATM, (a) wide scan spectra; (b) spectra of As 3d electrons; (c) spectra of Mn 2p electrons; and (d) spectra of O 1s electrons (virgin ATM = ATM, ATM after adsorption of As(III) = (ATM + As(III)) and ATM after adsorption of As(V) = (ATM + As(III))). | |
 |
| | Fig. 7 FTIR spectra of virgin ATM and after interaction with As(III), and As(V), (M* = Al, Ti, Mn). | |
Table 4 Population of O2− oxygen and –OH groups obtained from fitted XPS spectra of the O 1s peak of the optimized ATM before and after As(III) and As(V) adsorption
| Sample |
Peak |
FWHM, eV |
Percentage, % |
| ATM |
O2− |
2.6 |
52 |
| OH− |
2.6 |
48 |
| ATM + As(III) |
O2− |
2.7 |
61 |
| OH− |
2.7 |
39 |
| ATM + As(V) |
O2− |
2.5 |
56 |
| OH− |
2.5 |
44 |
The FTIR spectrum of ATM has strong hydroxyl stretching (3337 cm−1) and bending (1634 cm−1) vibrations of physically adsorbed H2O71,72 (Fig. 7 and S4†). The bands at 1111, 1071 and 978 cm−1 generally correspond to the bending vibration of hydroxyl groups of metal oxides (M*–OH).26,73 After adsorption of As(III) and As(V), the three peaks of hydroxyl groups weakened significantly, and new bands, corresponding to As–O stretching vibration, appeared at 803 cm−1 and 811 cm−1 for As(III) and As(V) adsorption experiment, respectively.74,75
According to Pena et al., the values of asymmetric stretching vibrations of (As–O) band position depend on the As(V) species present, which is strongly effected by the pH of the solution,42 and furthermore the intensity increases with the increase of As(V) concentration. The values ranged from 795 cm−1 in AsO43− to 909 cm−1 in H2AsO4− when the pH was decreased from 12.5 to 5.0.72 The corresponding values for adsorbed As(V) species were reported: 820 cm−1,76 828 cm−1,71 and 836 cm−1 (ref. 72) for the adsorption of As(V) on Fe–Mn oxide nanoparticles, Ce(IV)-doped iron oxide and Fe–Ce bimetal oxide, respectively. The shift of the band positions was attributed to a change in symmetry or asymmetry reduction originating directly from the formation of inner-sphere complexes on the Stern layer of surface of metal oxides adsorbent.
In our study, the As–O band position of adsorbed species appeared at 803 cm−1 and 811 cm−1 as a result of the formation of M*–O–As complexes.77 It is also in agreement with a previous FTIR study of As(V) adsorption on nanocrystalline titanium dioxide.42 These results indicate that the adsorption process of arsenic on the ATM adsorbent was completed through formation of a surface complex by substituting hydroxyl with arsenic species. That is the pre-dominant mechanism for As(V) adsorption on the ATM.
TGA-DTA analyses of ATM, TM, M, ATM + water and ATM after adsorption of As(III) and As(V) are shown in Fig. S6.† The experimental results of thermogravimetric analysis of Ti–Mn binary hydrous oxide (TM) and manganese hydrous oxide (M) in Fig. S6b and S6c,† respectively, confirmed the reference values in ref. 78 and 79 that the surface of composites containing Mn-oxides reach the surface state free of –OH group at T2 = 800 °C. Therefore, the TGA-DTA data of all the samples were evaluated in the range of temperature from T1 120 °C to T2 = 800 °C. According to the analyses and results shown in Fig. S6a1† and Table 5, the ATM has a high concentration of surface hydroxyl (125.39 × 1020 OH per g) and OH density (DOH, 162.624 OH per nm2). The DOH is by two orders of magnitude higher than for the other types of binary metal oxides.80,81 The concentration of surface hydroxyl of the sample ATM was reduced from 125.39 × 1020 OH per g to 112.39 × 1020 OH per g after shaking in distilled water and drying at room temperature. The decrease may be caused by the reaction between hydroxyl groups on the ATM surface and H+ in distilled water. When the blank sample (ATM + water) is compared with ATM samples after interaction with the AS(III) and As(V) in terms of concentration of surface hydroxyl, it is evident that hydroxyl groups were consumed due to the formation of complexes between M*–OH on ATM surface and the As(V) species, involving one or two surface hydroxyl with anion species:82
| | |
M*–OH + L = S–L+ + OH−
| (7) |
| | |
2M*–OH + L = 2S–L+ + 2OH−
| (8) |
where M* stands for aluminum, titanium and manganese, L stands for arsenic species.
Table 5 Surface hydroxyl (–OH) group determination for ATM, Ti–Mn binary hydrous oxide (TM), ATM after shaking in water and ATM after adsorption of As(III) and As(V). The specific surface area of ATM and TM is 71 and 336 m2 g−1, respectively
| Sample |
DOH (OH per nm2) |
OH × 1020 (OH per g) |
| ATM |
162.64 |
125.39 |
| TM |
17.39 |
58.25 |
| ATM + water |
145.78 |
112.39 |
| ATM + As(III) |
98.58 |
76.01 |
| ATM + As(V) |
106.96 |
82.46 |
The density of the hydroxyl groups dropped by 30% due to the adsorption of arsenic species. Interestingly, the addition of Al3+ in the synthesis process reduced the specific surface area of Ti–Mn binary hydrous oxide from 336 m2 g−1 to 71 m2 g−1, but it enhanced the number of –OH groups per square unit of ATM surface. Thus, the addition of Al3+ resulted in an increase in the –OH groups per gram of adsorbent, leading to an increase in adsorption capacity from 106.4 mg g−1, and 54.9 mg g−1 (TM) (Table S3, Fig. S5†) to 202.7 mg g−1, and 146.7 mg g−1 (ATM) for As(III) and As(V) at pH 7, respectively. The TGA analysis is the simplest method to prove that –OH groups on the ATM surface played an important role in As(III) and As(V) adsorption, which is consistent with the FTIR and XPS analyses.
4. Conclusions
A novel nanosized Al–Ti–Mn trimetal hydrous oxide was synthesized by a facile oxidation and co-precipitation method. Combining the features of single Al, Ti, Mn hydrous oxides, ATM exhibited an X-ray amorphous structure with a relatively large BET area (71 m2 g−1). The prepared ATM particles are aggregates formed by smaller nanosized particles. The adsorption kinetics and isotherm data were best fitted using the pseudo-second order model and the Langmuir model. The prepared trimetal hydrous oxide exhibited a high adsorption capacity towards both As(V) and As(III), being more effective in As(III) rather than As(V) removal. The maximal adsorption capacities for As(III) and As(V) are 202.7 mg g−1, and 146.7 mg g−1, at pH 7.0, which outperforms most reported adsorbents. The addition of Al3+ in the synthesis process facilitates an increase in the number of M*–OH active sites on the surface of the ATM absorbent, resulting in higher adsorption capacity, which is controlled by the hydroxyl exchange mechanism. Mn-hydrous oxide acted as oxidant ensuring complete conversion of As(III) to As(V) before the adsorption. Due to its excellent arsenic removal performance as well as simple and low-cost synthesis process, the ATM could be a promising adsorbent for both As(III) and As(V) removal from aqueous solutions.
Acknowledgements
The publication is a result of the project Development of the UniCRE Centre (project code LO1606) – “Green synthesis”, which was financially supported by the Ministry of Education, Youth and Sports of the Czech Republic under the National Programme for Sustainability.
References
- P. L. Smedley and D. G. Kinniburgh, Appl. Geochem., 2002, 17, 517–568 CrossRef CAS.
- P. Ravenscroft, H. Brammer and K. Richards, Arsenic Pollution: A Global Synthesis, Wiley, 2011 Search PubMed.
- L. Winkel, M. Berg, M. Amini, S. J. Hug and C. Annette Johnson, Nat. Geosci., 2008, 1, 536–542 CrossRef CAS.
- W. H. Organization, WHO web site, http://www.who.int, Accessed, 2012, 20.
- K. Straif, L. Benbrahim-Tallaa, R. Baan, Y. Grosse, B. Secretan, F. El Ghissassi, V. Bouvard, N. Guha, C. Freeman, L. Galichet and V. Cogliano, Lancet Oncol., 2009, 10, 453–454 CrossRef PubMed.
- N. Yamato, Bull. Environ. Contam. Toxicol., 1988, 40, 633–640 CrossRef CAS PubMed.
- M. F. Hughes, L. M. Del Razo and E. M. Kenyon, Toxicology, 2000, 143, 155–166 CrossRef CAS PubMed.
- D. Mohan and J. C. U. Pittman, J. Hazard. Mater., 2007, 142, 1–53 CrossRef CAS PubMed.
- B. A. Manning, S. E. Fendorf, B. Bostick and D. L. Suarez, Environ. Sci. Technol., 2002, 36, 976–981 CrossRef CAS PubMed.
- S. K. Maji, A. Pal and T. Pal, J. Hazard. Mater., 2008, 151, 811–820 CrossRef CAS PubMed.
- R. V. Hedegaard and J. J. Sloth, Biotechnol., Agron., Soc. Environ., 2011, 15, 45 Search PubMed.
- F. Bertolero, G. Pozzi, E. Sabbioni and U. Saffiotti, Carcinogenesis, 1987, 8, 803–808 CrossRef CAS PubMed.
- A. B. Ribeiro, E. P. Mateus and N. Couto, Electrokinetics Across Disciplines and Continents: New Strategies for Sustainable Development, Springer International Publishing, 2015 Search PubMed.
- V. Q. Chiu and J. G. Hering, Environ. Sci. Technol., 2000, 34, 2029–2034 CrossRef CAS.
- T. Nishimura and Y. Umetsu, Hydrometallurgy, 2001, 62, 83–92 CrossRef CAS.
- P. K. Dutta, S. O. Pehkonen, V. K. Sharma and A. K. Ray, Environ. Sci. Technol., 2005, 39, 1827–1834 CrossRef CAS PubMed.
- K. Gupta and U. C. Ghosh, J. Hazard. Mater., 2009, 161, 884–892 CrossRef CAS PubMed.
- S. Song, A. Lopez-Valdivieso, D. J. Hernandez-Campos, C. Peng, M. G. Monroy-Fernandez and I. Razo-Soto, Water Res., 2006, 40, 364–372 CrossRef CAS PubMed.
- A. Oehmen, R. Valerio, J. Llanos, J. Fradinho, S. Serra, M. A. M. Reis, J. G. Crespo and S. Velizarov, Sep. Purif. Technol., 2011, 83, 137–143 CrossRef.
- D. Nguyen Thanh, M. Singh, P. Ulbrich, N. Strnadova and F. Štěpánek, Sep. Purif. Technol., 2011, 82, 93–101 CrossRef.
- V. K. Sharma and M. Sohn, Environ. Int., 2009, 35, 743–759 CrossRef CAS PubMed.
- C. A. Martinson and K. J. Reddy, J. Colloid Interface Sci., 2009, 336, 406–411 CrossRef CAS PubMed.
- D. N. Thanh, M. Singh, P. Ulbrich, F. Štěpánek and N. Strnadová, Mater. Res. Bull., 2012, 47, 42–50 CrossRef CAS.
- M. Misono, Heterogeneous Catalysis of Mixed Oxides: Perovskite and Heteropoly Catalysts, Newnes, 2013 Search PubMed.
- S. K. Kulshreshtha, M. M. Gadgil and R. Sasikala, Catal. Lett., 1996, 37, 181–185 CrossRef CAS.
- G. Zhang, H. Liu, R. Liu and J. Qu, J. Hazard. Mater., 2009, 168, 820–825 CrossRef CAS PubMed.
- M. A. Barakat, Arabian J. Chem., 2011, 4, 361–377 CrossRef CAS.
- A. Maiti, S. DasGupta, J. K. Basu and S. De, Sep. Purif. Technol., 2007, 55, 350–359 CrossRef CAS.
- L. M. Camacho, R. R. Parra and S. Deng, J. Hazard. Mater., 2011, 189, 286–293 CrossRef CAS PubMed.
- K. R. Henke, Arsenic: environmental chemistry, health threats and waste treatment, Wiley, 2009 Search PubMed.
- V. Lenoble, C. Laclautre, B. Serpaud, V. Deluchat and J.-C. Bollinger, Sci. Total Environ., 2004, 326, 197–207 CrossRef CAS PubMed.
- Z. Orolínová and A. Mockovciaková, Mater. Chem. Phys., 2009, 114, 956–961 CrossRef.
- H. Y. Niu, J. M. Wang, Y. L. Shi, Y. Q. Cai and F. S. Wei, Microporous Mesoporous Mater., 2009, 122, 28–35 CrossRef CAS.
- T. Basu and U. C. Ghosh, J. Ind. Eng. Chem., 2011, 17, 834–844 CrossRef CAS.
- G. Zhang, J. Qu, H. Liu, R. Liu and R. Wu, Water Res., 2007, 41, 1921–1928 CrossRef CAS PubMed.
- Z. Li, S. Deng, G. Yu, J. Huang and V. C. Lim, Chem. Eng. J., 2010, 161, 106–113 CrossRef CAS.
- K. Wu, T. Liu, W. Xue and X. Wang, Chem. Eng. J., 2012, 192, 343–349 CrossRef CAS.
- K. Gupta, A. Maity and U. C. Ghosh, J. Hazard. Mater., 2010, 184, 832–842 CrossRef CAS PubMed.
- M. A. Ferguson, M. R. Hoffmann and J. G. Hering, Environ. Sci. Technol., 2005, 39, 1880–1886 CrossRef CAS PubMed.
- H. Zeng, B. Fisher and D. E. Giammar, Environ. Sci. Technol., 2007, 42, 147–152 CrossRef.
- J. Youngran, M. Fan, J. Van Leeuwen and J. F. Belczyk, J. Environ. Sci., 2007, 19, 910–919 CrossRef.
- M. Pena, X. Meng, G. P. Korfiatis and C. Jing, Environ. Sci. Technol., 2006, 40, 1257–1262 CrossRef CAS PubMed.
- D. G. Kinniburgh, J. K. Syers and M. L. Jackson, Soil Sci. Soc. Am. J., 1975, 39, 464–470 CrossRef CAS.
- R. W. M. Kwok, XPS Peak Fitting Program for WIN95/98 XPSPEAK version 4.1, 2000 Search PubMed.
- D. A. Shirley, Phys. Rev. B: Solid State, 1972, 5, 4709–4714 CrossRef.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- Y. S. Ho, J. C. Y. Ng and G. McKay, Sep. Sci. Technol., 2001, 36, 241–261 CrossRef CAS.
- P. Ratna Kumar, S. Chaudhari, K. C. Khilar and S. P. Mahajan, Chemosphere, 2004, 55, 1245–1252 CrossRef CAS PubMed.
- A. H. Welch and K. G. Stollenwerk, Arsenic in Ground Water, Springer, US, 2007 Search PubMed.
- U. C. Ghosh, D. Bandyopadhyay, B. Manna and M. Mandal, Water Qual. Res. J. Can., 2006, 41, 198–209 CAS.
- M. Singh, D. N. Thanh, P. Ulbrich, N. Strnadová and F. Stepánek, J. Solid State Chem., 2010, 183, 2979–2986 CrossRef CAS.
- J. Hlavay and K. Polyák, J. Colloid Interface Sci., 2005, 284, 71–77 CrossRef CAS PubMed.
- Z. Xu and X. Meng, J. Hazard. Mater., 2009, 168, 747–752 CrossRef CAS PubMed.
- K. Gupta, T. Basu and U. C. Ghosh, J. Chem. Eng. Data, 2009, 54, 2222–2228 CrossRef CAS.
- T. Wang, W. Yang, T. Song, C. Li, L. Zhang, H. Wang and L. Chai, RSC Adv., 2015, 5, 50011–50018 RSC.
- Z. Ren, G. Zhang and J. Paul Chen, J. Colloid Interface Sci., 2011, 358, 230–237 CrossRef CAS PubMed.
- W. Stumm and J. J. Morgan, Aquatic chemistry: chemical equilibria and rates in natural waters, Wiley, 1996 Search PubMed.
- Y.-M. Zheng, L. Yu, D. Wu and J. Paul Chen, Chem. Eng. J., 2012, 188, 15–22 CrossRef CAS.
- E. McCafferty and J. P. Wightman, Surf. Interface Anal., 1998, 26, 549–564 CrossRef CAS.
- L. V. Duong, B. J. Wood and J. T. Kloprogge, Mater. Lett., 2005, 59, 1932–1936 CrossRef CAS.
- J. F. Moulder and J. Chastain, Handbook of X-Ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data, Perkin-Elmer Corporation, Physical Electronics Division, 1992 Search PubMed.
- C. C. Surdu-Bob, S. O. Saied and J. L. Sullivan, Appl. Surf. Sci., 2001, 183, 126–136 CrossRef CAS.
- H. W. Nesbitt and D. Banerjee, Am. Mineral., 1998, 83, 305–315 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- G. C. Allen, S. J. Harris, J. A. Jutson and J. M. Dyke, Appl. Surf. Sci., 1989, 37, 111–134 CrossRef CAS.
- H. W. Nesbitt, G. W. Canning and G. M. Bancroft, Geochim. Cosmochim. Acta, 1998, 62, 2097–2110 CrossRef CAS.
- J. S. Foord, R. B. Jackman and G. C. Allen, Philos. Mag. A, 1984, 49, 657–663 CrossRef CAS.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1993, 20, 77–89 CrossRef CAS.
- M. J. Scott and J. J. Morgan, Environ. Sci. Technol., 1996, 30, 1990–1996 CrossRef CAS.
- L. Chen, B.-Y. He, S. He, T.-J. Wang, C.-L. Su and Y. Jin, Powder Technol., 2012, 227, 3–8 CrossRef CAS.
- Y. Zhang, M. Yang and X. Huang, Chemosphere, 2003, 51, 945–952 CrossRef CAS PubMed.
- Y. Zhang, M. Yang, X.-M. Dou, H. He and D.-S. Wang, Environ. Sci. Technol., 2005, 39, 7246–7253 CrossRef CAS PubMed.
- K. Nakamoto, Infrared and Raman spectra of inorganic and coordination compounds, Wiley, 1978 Search PubMed.
- S. C. B. Myneni, S. J. Traina, G. A. Waychunas and T. J. Logan, Geochim. Cosmochim. Acta, 1998, 62, 3499–3514 CrossRef CAS.
- S. C. B. Myneni, S. J. Traina, G. A. Waychunas and T. J. Logan, Geochim. Cosmochim. Acta, 1998, 62, 3285–3300 CrossRef CAS.
- B. An and D. Zhao, J. Hazard. Mater., 2012, 211–212, 332–341 CrossRef CAS PubMed.
- J. A. Tossell, Geochim. Cosmochim. Acta, 1997, 61, 1613–1623 CrossRef CAS.
- R. Mueller, H. K. Kammler, K. Wegner and S. E. Pratsinis, Langmuir, 2002, 19, 160–165 CrossRef.
- R. A. Lidin, L. L. Andreyeva and V. A. Molochko, Constants of Inorganic Substances: A Handbook, Begell House, 1995 Search PubMed.
- M. Chee Kimling, N. Scales, T. L. Hanley and R. A. Caruso, Environ. Sci. Technol., 2012, 46, 7913–7920 CrossRef CAS PubMed.
- D. Chen, L. Cao, T. L. Hanley and R. A. Caruso, Adv. Funct. Mater., 2012, 22, 1966–1971 CrossRef CAS.
- W. Stumm, Aquatic surface chemistry: chemical processes at the particle-water interface, Wiley, 1987 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra11347h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 









