
Synthesis of carbon nanoparticle embedded graphene for sensitive and selective determination of dopamine and ascorbic acid in biological fluids†
Sudip Biswasa,
Rashmita Dasa,
Malini Basua,
Rajib Bandyopadhyaya and
Panchanan Pramanik*b
aDepartment of Instrumentation and Electronics Engineering, Jadavpur University, Salt Lake Campus, Sector-III, Kolkata – 700098, India
bDepartment of Chemistry and Nanoscience, GLA University, Mathura – 281 406, India. E-mail: pramanik1946@gmail.com
First published on 27th September 2016
Abstract
We have prepared carbon nanoparticle embedded graphene (CNEG) by carbonizing a ternary composite of GO/melamine-formaldehyde resin/Zn(OAc)2. CNEG is characterized by TEM, FESEM, FTIR, Raman spectroscopy and BET. The CNEG showed narrow distributed pores with diameters from 1.55 to 7.40 nm and has a high surface area (241 m2 g−1). The CNEG is introduced as a substitute for graphite paste (GP) electrode with better performance. A CNEG modified GP (CNEG/GP) electrode shows better electrocatalytic oxidation towards ascorbic acid (AA), dopamine (DA) than both a bare GP electrode and a thermally reduced graphene oxide modified graphite paste (TRG/GP) electrode. Two well-separated voltammetry peaks are obtained using the CNEG/GP electrode in differential pulse voltammetry measurements and the corresponding peak separation between AA and DA is 192 mV facilitating simultaneous determination of these two biomolecules. Under optimized experimental condition, the linear ranges of determinations of DA and AA are found to be 0.07–200 μM and 25–2700 μM respectively. The lower detection limits for DA and AA are 50 nM and 520 nM respectively. Furthermore, the CNEG/GP electrode shows good reproducibility and chemical stability. It is also tested for the analysis of urine, serum and pharmaceutical products with better sensitivities.
1. Introduction
Dopamine (DA) is an important neurotransmitter in the central nervous system and acts as a local messenger in blood vessels, kidneys, the pancreas, and the digestive, cardiovascular and immune systems.1–3 Neurological disorders such as schizophrenia and Parkinson's disease may be induced by low levels of DA.4 It is used as an essential drug for low blood pressure, slow heart rate and cardiac arrest.5 Ascorbic acid (AA) is an important vitamin and a natural antioxidant.6,7 AA has an important role in collagen formation and in iron uptake.8 It is used as an auxiliary medicine for mental illness, infertility, common colds, cancer and AIDS. Deficiency of AA leads to anemia, deterioration of collagen, skin hemorrhages, lowering of body resistance from infections and is responsible for thyroid deficiency and premature aging.9–11 On the other hand, high levels of AA cause gastric irritation and diarrhea. Several analytical techniques for the determination of AA and DA have been developed using liquid chromatography,12 spectrophotometry,13 chemiluminescence, flow injection analytical methods,14 capillary electrophoresis,15 and colorimetry.16 But these methods are often expensive and time consuming with low sensitivity and selectivity. DA and AA are electro-active compounds; therefore electrochemical techniques have drawn a lot of attention in the last few years due to their simplicity for the simultaneous determination of AA and DA.17 Both of the above mentioned biomolecules do co-exist in biological fluids and possess close values of oxidation potentials. Thus, during electrochemical analysis of the mixtures, overlapping of the oxidation peaks occurs at conventional carbon electrodes. To overcome this problem, many attempts have been made to resolve the voltammograms using electrodes modified with polymer film,18 Au nanoparticle19 enzyme,20 CdTe nanoparticles,21 CNT composites,22–27 graphene and its composites28–35 etc. Out of these carbon materials, especially graphene and its composites, have received special attention due to its high work function, chemical inertness, high surface area, unique structures, outstanding charge-transfer characteristics. As a rising carbon material, it has great potential for the applications in many fields such as nanoelectronics, nanophotonics, catalysis, sensors, nanocomposites, supercapacitors due to its unique electronic, mechanical and thermal properties. So far graphene composite with Pt, Au, Pd, ZnO, CeO2, SnO2, TiO2, PANI, PPy have been employed extensively owing to their excellent electrocatalytic properties.Here first time we have synthesized a carbon nanoparticle embedded graphene (CNEG) by direct carbonization of a composite of melamine-formaldehyde resin (MFR) and GO (Scheme 1) with uniform dispersion of carbon nanoparticle on the graphene sheets with narrow pore size (1.55 to 7.40 nm) distribution with high surface area(241 m2 g−1). The CNEG modified GP (CNEG/GP) electrode has shown high electrocatalytic activity towards AA and DA. Here carbon nanoparticles doped with nitrogen act as an absorber for biomolecules. Also, we have compared the analytical results with previously reported work, and the present electrode found to be superior to all reported methods (Table 1) in the analysis of mixture of DA and AA in various systems including biological fluids.
| Electrode materials | Linear range (μM) | Detection limit (μM) | ||
|---|---|---|---|---|
| AA | DA | AA | DA | |
| a NDG: nitrogen doped graphene PtNPs/MWCNT: Pt nanoparticles decorated multi wall carbon nanotube. PtNCs/graphene: platinum nanocomposites/graphene. | ||||
| Au@Pd–graphene47 | 0.1–1000 | 0.01–100 | 0.002 | 0.02 |
| Graphene/Au NPs34 | — | 5–1000 | — | 1.6 |
| NDG38 | 5–1300 | 0.5–170 | 2.2 | 0.25 |
| Pd NPs/chitosan–graphene30 | 100–4000 | 0.5–15, 20–200 | 20 | 0.1 |
| Pt NPs/MWCNT31 | 24.5–765 | 0.061–2.03 | 20 | 0.048 |
| Pd–Pt/graphene48 | 40–1200 | 4–200 | 0.61 | 0.04 |
| PtNCs/graphene32 | 0.15–34.4 | 0.03–8.13 | 0.15 | 0.03 |
| Pt–Au hybrid49 | 103–165 | 24–384 | 100 | 21 |
| rGO/TiO2 (ref. 50) | 400–3000 | 2–200 | 200 | 6 |
| Fe3O4/graphene51 | 1000–9000 | 0.5–100 | 0.42 | 0.12 |
| SnO2/graphene52 | — | 2–100 | — | 2 |
| CNEG/GP (this wok) | 25–1000–2700 | 0.07–0.3–200 | 0.520 | 0.05 |
2. Experimental
2.1 Chemicals and reagents
Formaldehyde, zinc acetate dihydrate, ascorbic acid, dopamine, sodium dihydrogen phosphate, sodium hydroxide, and paraffin oil were obtained from Merck, India. Melamine was purchased from Sigma Aldrich, USA. Graphite flakes were from SRL, India. Dopamine injection and ascorbic acid tablet are purchased from the local drug store. All the reagents were analytical graded and used without further purification. All required solutions were prepared using Millipore water (resistance 18 MΩ).2.2 Apparatus and measurements
The morphology of synthesized CNEG was determined by JEM-2100 high resolution transmission electron microscope (HRTEM) and JEOL-JEM-6700F field emission scanning electron microscopy (FESEM). The TEM sample was prepared by drying a droplet of suspended CNEG on a carbon coated Cu grid. Nitrogen adsorption–desorption isotherms and Brunauer–Emmett–Teller (BET) surface areas were measured by using Beckmam Coulter SA3100 at 77 K. The sample was degassed at 200 °C for 3 h. Powder X-ray diffraction (PXRD) was carried out with an X-ray diffractometer (XRD, D/MAX-RA, Japan) operated at 40 kV and 40 mA with CuKα radiation (λ = 0.15406 nm). Fourier transform infrared (FTIR) spectra (KBr dispersed pellets) in the range of 400–4000 cm−1 were obtained from the Perkin Elmer FTIR spectrometer (model Paragon-500 FTIR). The nitrogen content in CNEG was determined using an elemental analyzer (Vario EL III, Germany). All electrochemical measurements were performed by three electrode system using Autolab Potentiostate/Galvanostate 101 (Netherlands). An Ag/AgCl electrode was used as a reference electrode and Pt electrode as a counter electrode. All the electrochemical studies were carried out in 0.1 (M) phosphate buffer of pH 6 at temperature 25 ± 2 °C. The real samples were collected by the members of our laboratory and cross-analyses were done by a standard pathological laboratory. All the analyses were performed in compliance as per the guidelines of the institute and with the consent for the experimentation with human subjects.2.3 Synthesis of CNEG
Graphene oxide (GO) is synthesized by modified Hummer's method.36 CNEG was synthesized by first dispersing 500 mg of GO in 300 ml of Millipore water with 1 g of Zn(OAc)2, 2H2O and sonicating for one hour. Then, 1.25 g of melamine was added to the above mixture. After that 4.5 ml of formaldehyde was added drop wise with continuous stirring at 80 °C until evaporated to dryness. After that, the dried solid powder was carbonized in a horizontal tube furnace at 950 °C for 1 h with a heating rate of 5 °C min−1 under the flow of N2 gas. The obtained black powder was collected. Complete removal of Zn species is confirmed by energy-dispersive X-ray spectroscopy (EDS) in Fig. 2d.2.4 Preparation of CNEG/GP electrode
Graphite flakes and synthesized CNEG were mixed in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w) ratio in a mortar and paraffin oil was added, followed by grinding for making a paste. Finally, the paste was poured into a capillary glass tube of 2 mm inner diameter and packed tightly by pressing with a metal rod and a hydraulic press. Electrical connection was done by putting a Cu-wire from the backside of the glass tube. Bare graphite and TRG/GP paste electrodes were prepared by similar method. Before experimentation, electrode surface was cleaned with 0.3 μm and 0.05 μm Al2O3 slurries, and rinsed with ethanol and dried under N2.
:1 (w/w) ratio in a mortar and paraffin oil was added, followed by grinding for making a paste. Finally, the paste was poured into a capillary glass tube of 2 mm inner diameter and packed tightly by pressing with a metal rod and a hydraulic press. Electrical connection was done by putting a Cu-wire from the backside of the glass tube. Bare graphite and TRG/GP paste electrodes were prepared by similar method. Before experimentation, electrode surface was cleaned with 0.3 μm and 0.05 μm Al2O3 slurries, and rinsed with ethanol and dried under N2.
3. Results and discussion
3.1 Formation mechanism of NDPC
Scheme 1 is showing a graphical representation of the formation of CNEG, which is synthesized through two major steps. In the 1st step melamine is reacted with formaldehyde in the presence of Zn(OAc)2 to form Zn(OAc)2 incorporated melamine-formaldehyde resin (MFR) at 80 °C and eventually is grafted on GO sheets to form a ternary composite, GO/MFR/Zn(OAc)2. In the 2nd step during annealing at 950 °C, MFR on GO sheet decomposes to carbon nanoparticle and GO is converted to graphene at high temperature. During high temperature thermal annealing all the Zn species reduced to metallic zinc (b.p 906 °C) and evaporated out from the sample to make a porous structure.37,38 Here Zn(OAc)2 is used as an activator and a pore forming agent. The nitrogen content in the CNEG was measured by EDS and was found to be 5.21 atomic wt. percentages (Fig. 2d, see ESI Table S1†). | ||
| Scheme 1 Illustration of formation of CNEG, in (step-1) formation of GO/MFR/Zn(OAc)2 composite, (step-2) removal of Zn species with formation of the CNEG. | ||
3.2 XRD and BET analysis of prepared CNEG
The powder X-ray diffraction (PXRD) studies of carbon nanoparticle embedded graphene (CNEG) and GO/MFR/Zn(OAc)2 have been carried out and presented in Fig. 1a and b. PXRD of GO/MFR/Zn(OAc)2 (Fig. 1a) shows well define (002) graphitic plane at 2θ = 23.3° for fully oxidized graphite with several peaks for ZnO. Whereas, PXRD of CNEG in Fig. 1b shows two well define graphitic peak at 2θ = 26.30° for (002) plane and 43.30° for (101) plane. Here, the positive shift of (002) peak is due to thermal reduction of GO to reduced graphene. There is no peak for Zn species. This may explain as, at higher temperature (>800 °C) all the ZnO have been converted to metallic zinc, which eventually evaporated out at a temperature higher than 906 °C.37 Also the complete removal of zinc species is being confirmed by energy dispersive X-ray spectroscopy (Fig. 2d and see ESI Table S1†). | ||
| Fig. 1 PXRD of (a) GO/MFR/Zn(OAc)2 and (b) CNEG, (c) nitrogen adsorption–desorption isotherms (black and red symbols represent adsorption and desorption isotherms, respectively) and (d) corresponding BJH pore size distributions of the CNEG. | ||
Nitrogen adsorption/desorption isotherms at 77 K and pore size peak distribution plot was calculated by BJH (Barrett, Joyner and Halenda), adsorption measurement within a relative pressure range from 0.01 to 101.3 kPa. The samples were exposed to gas mixture of He and N2 with a programmed ratio, adsorbed amount of N2 was calculated from the change of pressure in the cell after equilibrium. Before nitrogen adsorption/desorption measurements, the samples were degassed at 200 °C under vacuum for 3 h.
The N2 adsorption–desorption isotherm presented in Fig. 1c shows a reversible adsorptions character which is like type-II isotherm. It provides evidence for micropores with surface area 241 m2 g−1 which is lower than the theoretical surface area (2630 m2 g−1) for individually isolated graphene sheet due to the formation of stacks of multi-layered graphene.
However BJB pore diameter distribution spread over a range from 1.55 to 7.40 nm (Fig. 1d) shows that the obtained CNEG is mesoporous cum microporous in nature. Pore diameter larger than 2 nm in diameter is suitable for quick mass transfer of molecules and ion diffusion in aqueous electrolyte. Furthermore, CNEG has a small pore diameter, high surface area, nitrogen doping with embedded carbon nanoparticles and is expected to have good electrocatalytic properties.
3.3 FESEM and TEM of GO/MFR/Zn(OAc)2 and CNEG
The morphology of the GO/MFR/Zn(OAc)2 and CNEG were studied by field emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM) and represented in Fig. 2a–c. The FESEM of GO/MFR/Zn(OAc)2 shows the incorporation of MFR into the GO layers. In the TEM image shows the dispersion of uniform sized carbon nanoparticles on the graphene layer inset Fig. 2c. Also inset SAED patterns in Fig. 2c also present a ring like pattern derived from typical amorphous carbon, which is in agreement with the XRD results mentioned above. Fig. 2d shows energy-dispersive X-ray spectroscopy (EDS) of CNEG. The nitrogen doping is found to be 5.12 atomic weight percentages.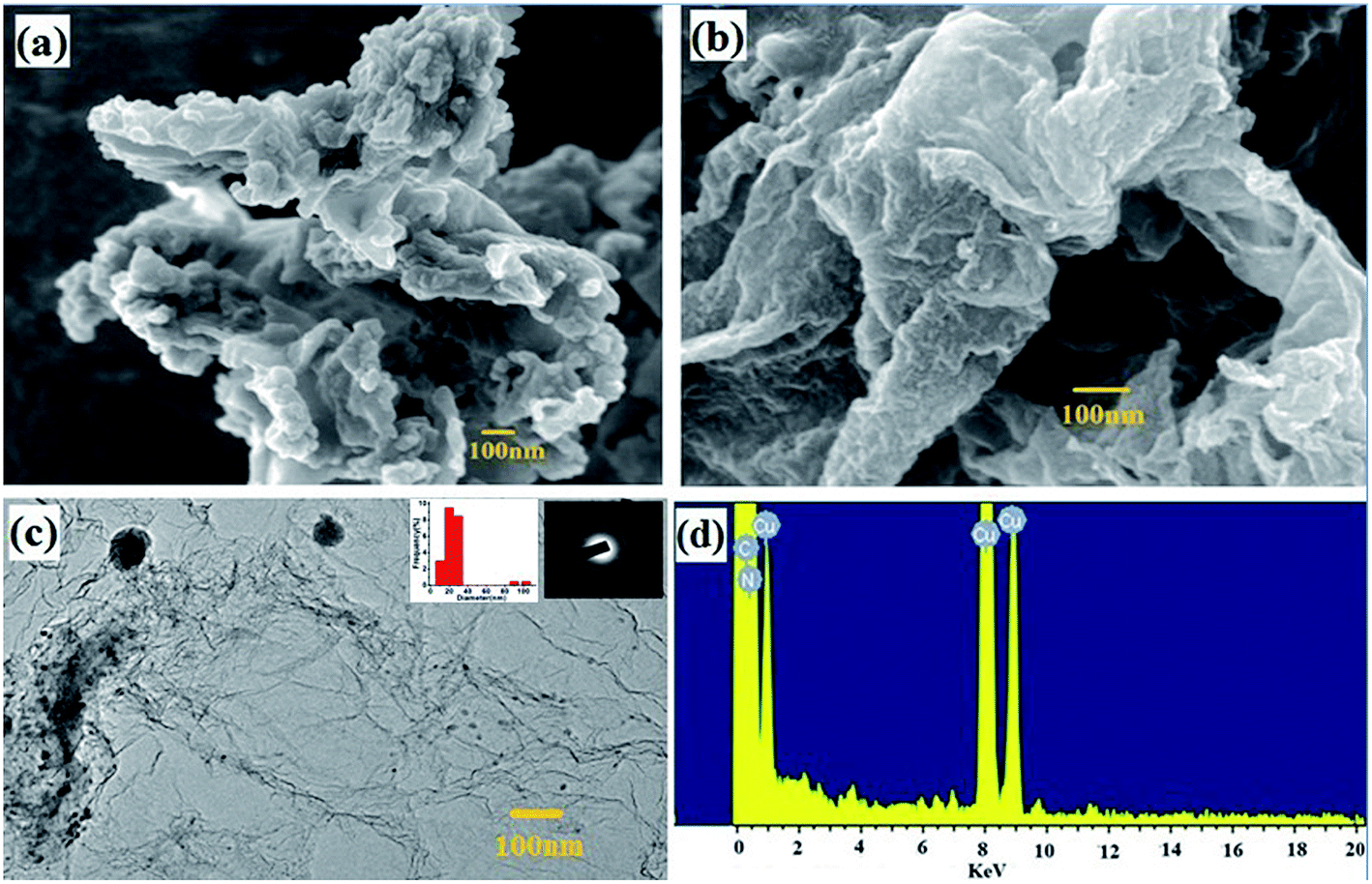 | ||
| Fig. 2 (a) FESEM image of GO/MFR/Zn(OAc)2, (b) and (c) FESEM and TEM image of CNEG (inset particle size distribution and SAED) (d) EDS spectra of CNEG. | ||
3.4 FTIR study of GO/MFR/Zn(OAc)2 and CNEG
Fourier transform infrared (FTIR) spectroscopy is carried out with GO/MFR/Zn(OAc)2 composite (red) sample and also after pyrolysis of CNEG (black) and are presented in Fig. 3. The FTIR spectra of GO/MFR/Zn(OAc)2 contains overlapped signature of the individual component of the composite. The stretching of O–H overlaps with the bending vibration of NH2 in MFR, resulting in a broad peak from 3105 to 3455 cm−1. The two weak peaks at 2850 and 2933 cm−1 indicate stretching bands of C–H. The peak at 1382 cm−1 is related to the C–N stretching of the secondary/tertiary amine group. The FTIR peaks at 814, 1002 and 1150 cm−1 correspond to C–N–C, ether(C–O–C) and the amino group respectively. The triazine unit in MFR shows two peaks at 1570 and 1470 cm−1. A band at1590-1620 cm−1 is generated from unoxidized C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (sp2). The peak at 476 cm−1 may be due the presence of ZnO. During pyrolysis of MFR, rupture of triazine units takes place between 600 °C to 675 °C (ref. 39) and above 906 °C, all the metallic Zn evaporates. The mutual effects of these two lead to the formation of porous CNEG. The FTIR of CNEG indicates the presence of graphitic structure through broad peak between 1000 cm−1 and 4000 cm−1 with C–N stretching at 1382 cm−1.
C (sp2). The peak at 476 cm−1 may be due the presence of ZnO. During pyrolysis of MFR, rupture of triazine units takes place between 600 °C to 675 °C (ref. 39) and above 906 °C, all the metallic Zn evaporates. The mutual effects of these two lead to the formation of porous CNEG. The FTIR of CNEG indicates the presence of graphitic structure through broad peak between 1000 cm−1 and 4000 cm−1 with C–N stretching at 1382 cm−1.
 | ||
| Fig. 3 FTIR spectra of GO/MFR/Zn(OAc)2 composite (red) and CNEG (black). | ||
3.5 Electrochemical properties study of CNEG/GP, bare GP and TRG/GP electrodes
A standard [Fe(CN)6]4−/[Fe(CN)6]3− redox system is used to study the electrocatalytic properties of the electrodes. The cyclic voltammograms obtained for 3 mM K4[Fe(CN)6] solution using bare GP, CNEG/GP and TRG/GP electrodes are presented in Fig. 4a. The peak potential separation (ΔEp) at CNEG/GP electrode is 88 mV and that for bare GP and TRG/GP electrodes are 391 mV and 108 mV respectively. According to Valasco equation, electron transfer rate increases with a decrease in the value of ΔEp which indicates that the electron transfer rates are in the order of CNEG/GP > TRG/GP > bare GP electrode. The redox peak current is highest for the CNEG/GP electrode; this may arise due to large surface area and good electrical conductivity. All these results show that CNEG/GP is significantly better than bare GP and TRG/GP electrode. The CNEG/GP electrode shows better sensitivity at pH 6 (Fig. 4b), therefore all the analytical work is carried out at this optimum pH. | ||
| Fig. 4 (a) CVs obtained for 3 mM Fe(CN)64−/Fe(CN)63− at bare GP (black), CNEG/GP (red), and TRG/GP(blue) electrode (b) pH vs. peak current (IAA/DA) of 0.1 mM individual AA and DA at CNEG/GP electrode. Scan rate for CVs: 50 mV s−1. | ||
3.6 Electrocatalytic oxidation of AA and DA
The redox studies of individual as well as a mixture of AA and DA are studied by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) at bare GP, TRG/GP and CNEG/GP (Fig. 5a and b). Fig. 5a shows CVs of AA at different electrode. It was documented that oxidation of anionic AA (pKa = 4.17) require one proton and two electrons transfer.40 It can be seen that AA is oxidized irreversibly at all three electrodes. AA is oxidized at bare GP and TRG/GP electrode with a large over potential indicating sluggish electron transfer may caused by fouling effect of oxidation product of AA.41 AA shows a negative shift of oxidation potential at CNEG/GP (−2 mV) from those of TRG/GP (286 mV) and bare GP (315 mV) by 288 mV and 317 mV respectively. Furthermore, the oxidation peak current at CNEG/GP (2.95 μA) is 3.8 and 1.3 time higher than that of bare GP (0.768 μA) and TRG/GP (2.3 μA) respectively. The CVs (see ESI, Fig. S1a†) of AA oxidation at the CNEG/GP electrode with varying scan rates ranging from 5 to 400 mV s−1. The inset plot shows oxidation peak current is linear with scan rate which indicates the electrochemical oxidation of AA is a diffusion controlled process at the CNEG/GP electrode. | ||
| Fig. 5 CVs of at bare GP (black), CNEG/GP (red) and TRG/GP (red) electrodes of (a) 0.1 mM AA; (b) 0.1 mM DA and (c) 0.1 mM AA + 0.1 mM DA mixture in 0.1 (M) phosphate buffer pH 6.0; DPV of (d) 0.1 mM of AA + 0.1 mM of DA mixture at CNEG/GP electrode in 0.1 (M) phosphate buffer pH 6.0. Scan rate for CVs: 50 mV s−1 and for DPVs: 8 mV s−1. | ||
CVs of DA are represented in Fig. 5b. The redox nature of DA has been documented to be the process of transfer of two-electrons and two-protons. The oxidation peak is arisen due to the oxidation of dopamine to dopaminequinone, while the reduction peak is from the reverse phenomenon.42 DA is shown a quasi-reversible redox property with redox peak separation (ΔEp) of 85 mV and 44 mV at bare GP and TRG/GP electrode respectively, however in case of CNEG/GP it becomes only 32 mV which implies better reversibility. Meanwhile, the peak current value at CNEG/GP electrode (3.74 μA) is 3.74 times and 1.33 times higher than those at bare GP (1 μA) and TRG/GP (2.80 μA) electrodes, respectively. However, the increase of anodic peak current with increasing scan rate is proportional within the range of 5 to 300 mV s−1 (see ESI, Fig. S1b†), demonstrating a diffusion controlled process.
CVs of a mixture of AA and DA at different electrode are represented in Fig. 5c. At bare GP and TRG/GP electrodes, the oxidation peaks of AA and DA are overlapped with each other which results a board oxidation peak. In contrast, at CNEG/GP electrode the oxidation peaks are well resolved with an oxidation peak potential at 93 mV and 271 mV for AA and DA respectively. The peak potential separation is 181 mV and 192 mV for AA-DA by CV and DPV respectively (Fig. 5d). The peak current value for DA (6.3 μA) oxidation is much higher than AA (3.3 μA) for a mixture of 0.1 mM of each. The results indicate that CNEG/GP has better electrocatalytic activities towards the oxidation of AA and DA. The combination of carbon nanoparticles and graphene enhanced the electronic conductivity with biomolecules accessibility. Which facile the electron transfer rate between the biomolecules and electrode surface and thereby caused a good synergistic effect in the electrocatalysis. Also CNEG/GP has the potentials to resolve the oxidation peak of AA and DA in mixture. The tentative mechanisms may be as follows:
(1) The nitrogen atoms in CNEG may interact with AA or DA via hydrogen bonding, which can activate the hydroxyl or amine groups and accelerate the charge transfer kinetics at CNEG/GP electrode.39
(2) CNEG has high surface area and narrow pore diameter distribution between 1.4 to 7.4 nm, which are suitable for faster mass transfer of the biomolecules in aqueous electrolyte.43,44
(3) Due to the N-doping in CNEG, re-orientations of the energy levels take place (see ESI, Fig. S2†).
(4) Carbon nanoparticles increase the adsorption property.45 Due to π–π interaction or H-bonding with the biomolecules at nitrogen doped carbon nanoparticle.
The excellent electrocatalytic activities of CNEG may be due to their mesoporous structure and catalytic properties are originating from the adsorptive carbon nanoparticles along with the N-doping effect. All these phenomena have introduced simultaneous determinations of AA and DA more efficiently at the CNEG/GP electrode.
3.7 Simultaneous determination of AA and DA
Differential pulse voltammetry (DPV) has a better current sensitivity and higher resolution over CV by minimizing non-faradic current, therefore all the analytical works are carried out using a DPV. Fig. 6 shows the simultaneous determination of AA and DA in a mixture. DPV curves are obtained by changing the concentration of one constituent while keeping the other at a constant value. The oxidation peak currents of AA and DA increase linearly with increase of the concentrations of DA and AA. The oxidation peak currents for AA shown a linear relationship with the AA concentration (Fig. 6a) from 25 to 1000 μM with the linear regression equation is calibrated as IAA (μA) = 0.015CAA (μM) + 0.55 with the correlation co-efficient of R2 = 0.970 and again 1000 μM to 2700 μM with the linear regression equation IAA (μA) = 0.0043CAA (μM) + 12.26 with the correlation co-efficient of R2 = 0.980. In Fig. 6b, the oxidation peak currents of DA increases with the increase of DA concentrations (Fig. 6b), 1st from 0.07 μM to 0.3 μM and then 0.5 μM to 200 μM, the corresponding linear equations are IDA (μA) = 1.65CDA (μM) + 0.0234 (R2 = 0.999) and IDA (μA) = 0.0401CDA (μM) + 0.428 (R2 = 0.990) for the respective linier range. The measured limits of detection for AA and DA are 430 nM, 50 nM respectively. | ||
| Fig. 6 DPVs at CNEG/GP electrode in 0.1 (M) PBS (pH 6.0) at CNEG/GP electrode (a) containing 0.1 mM DA and different concentrations of AA: 25, 50, 100, 200, 400, 800, 1000, 1500, 2000, 2500, 2700 μM; (inset) corresponding peak current (IAA) vs. concentration (CAA) curve; (b) containing 0.1 mM AA and different concentrations of DA: 0.07, 0.1, 0.3, 0.5, 1, 10, 20, 30, 50, 70, 120, 140, 180, 200, 210 μM; (inset) corresponding concentration (CDA) vs. peak current (IDA) curve; (c) electron and hole transfer during oxidation of the biomolecules. (d) Schematic of the adsorbed biomolecule. DPV conditions: a pulse height of 50 mV, a step height of 4 mV, a pulse width of 0.2 s, a step time of 0.5 s, and a scan rate of 8 mV s−1. | ||
These results could be explained as follows.
(1) Peak current: DA contain an aromatic ring in their molecular structure, resulting in strong adsorption on the carbon surface, introducing high current for oxidation. The oxidation peak current is a mutual effect of the electron flow to the CNEG/GP (Fig. 6c) and the adsorption strength (Fig. 6d). The m-LOD reached 50 nM for DA. However, the adsorption strength of AA was weak due to the absence of an aromatic ring. As a consequence, the oxidation current was low compared to DA, resulting in a relatively poor detection limit.
(2) Linear range: the existence of two slopes in the oxidation peak current may be explained by monolayer adsorption followed by multilayer adsorption.46
(3) Oxidation potential: the Schottky barrier that forms at the CNEG/GP and biomolecule interface is the main origin of the oxidation potential but it also depends on electrolyte acidity, scan rate and adsorption of biomolecule at different concentrations. Here, after a particular concentration of biomolecules brings the shift of oxidation potential due to dominant adsorption of biomolecule on the electrode surface.
Even at low concentrations of DA, the first layer was easily formed due to strong adsorption. In the limit of a high concentration, interactions between the second-layer molecules were screened by the first-layer molecule, which means that the interaction strength differs from that of the first-layer molecules, resulting in two slopes in the peak current. Due to the relatively weak interaction of the AA molecules, multilayer adsorption of the molecules is realized at very high concentrations. Also DA had a higher slope than AA, which is reflected as a higher sensitivity.
Due to the coexistence of DA and AA in numerous biological systems, accurate measurements of each component with a high degree of sensitivity and selectivity are mandatory. This investigation has established that the nano-composite of graphene with nitrogen doped amorphous carbon nanoparticle has excellent performance for simultaneous determination of the amount of DA and AA in a binary mixture without mutual interferences. The detection limit is very low in comparison to earlier reported literatures and selectivity is quite high. These analytical results have implied that the simultaneous determination of AA and DA can be achieved with high sensitivity and selectivity at the CNEG/GP electrode. Furthermore, the present results are compared with other reported works (Table 1). We can notice that the analytical performances, limit and linear range of the CNEG/GP electrode are better than or comparable to the results reported previously for simultaneous determination of these two biomolecules. To the best of our knowledge, such low detection limits and linear range for direct electrochemical determination of AA and DA in the coexistence system of AA and DA are rarely reported.
3.8 Reproducibility and stability
To find the reproducibility and stability of CNEG/GP electrode, the peak currents of DPV in a binary mixture solution containing 0.1 mM AA, 0.1 M DA was recorded for 5 successive measurements. The relative standard deviations (R.S.D.) are 2.1% and 1.2% for AA and DA respectively. The reproducibility of CNEG/GP electrodes is carried out in the same solution mixture and the R.S.D. (n = 3) values are found to be 3.10%, 2.95% for AA and DA respectively. Meanwhile, CNEG/GP electrode is stored in air at room temperature for three weeks; the CNEG/GP electrode shows no obvious change in the analytical performance (change in peak current values of 1.2%, 2.1% for AA and DA respectively). All these results demonstrate that the CNEG/GP electrode has good stability and reproducibility.3.9 Interference study
The interferences of different substrates during the determination of AA and DA at CNEG/GP are studied under optimum experimental condition (50 μM AA + 50 μM DA in 0.1 (M) PBS at pH 6). The commonly interfering agents such as glucose, sucrose, citric acid, ethanol, Na+, K+, Mg2+, SO42−, NH4+, Fe2+, Fe3+, CO32−, Cl−, uric acid, H2O2, adenine, folic acid, and urea. The results show that there is no significant change of current for the simultaneous determination of AA and DA i.e. error is less than ±0.7 μA.3.10 Real sample analysis
For a real sample analysis, we have taken urine, cerebrospinal fluid and serum. The real samples are diluted with 0.1 (M) phosphate buffer (pH 6) to an appropriate level, i.e. 40 times for urine sample and 2 times in the serum and cerebrospinal fluid sample, to fit the calibration curve. Dopamine hydrochloride injection is diluted 20 times with 0.1 (M) phosphate buffer (pH 6) and another solution of AA is prepared by grinding ascorbic acid tablet. No other pretreatment process was performed. The results obtained for real samples are summarized in ESI (see ESI, Tables S2, S3 and Fig. S3–S6†). The recovery values for AA and DA in urine sample are 100.2% and 101.5% respectively and in serum sample are 102.5% and 100.8%. All these results are indicating that the CNEG/GP electrode has greater potential for the simultaneously determine of AA and DA in real samples.4. Conclusions
We have synthesized carbon CNEG by direct carbonization of a ternary composite of GO/MFR/Zn(OAc)2 and used for simultaneous determination of AA and DA. Here we have used Zn(OAc)2 as an activator and pore forming agent. Due to the narrow pore-size distribution, high surface area, nitrogen-doping effect and the presence of carbon nanoparticles on graphene surface have introduced better electrochemical performances for the oxidation of AA and DA compared to the TRG/GP and bare GP electrodes.Acknowledgements
The authors are grateful to Council of Scientific and Industrial Research (CSIR) India for financial support for this work. We also thank to UGC-DAE consortium for scientific research, Kolkata centre for XRD study.Notes and references
- M. B. Murphy, J. Hum. Hypertens., 2000, 1, S47–S50 Search PubMed.
- H. Leblanc, G. C. Lachelin, S. Abu-Fadil and S. S. Yen, J. Clin. Endocrinol. Metab., 1976, 43, 668–674 CrossRef CAS PubMed.
- S. Li and G. Pelletier, Endocrinology, 2013, 131, 395–399 Search PubMed.
- J. Birtwistle and D. Baldwin, Br. J. Nurs., 1998, 7, 832–841 CrossRef CAS PubMed.
- A. Miles, A. Shulan and J. W. Cheng, Ann. Pharmacother., 2013, 47, e21 CrossRef PubMed.
- O. Arrigoni and M. C. De Tullio, Biochim. Biophys. Acta, 2002, 1569, 1–9 CrossRef CAS.
- M. C. De Tullio, Subcell. Biochem., 2012, 56, 49–65 CAS.
- L. Hallberg, M. Brune and L. Rossander, Int. J. Vitam. Nutr. Res., Suppl., 1989, 30, 103–108 CAS.
- J. Thomson, M. Manore and L. Vaughan, The Science of Nutrition, Benjamin Cummings, San Francisco, CA, 3rd edn, 2013, ch. 10 Search PubMed.
- K. A. Naidu, Nutr. J., 2003, 2, 7 CrossRef PubMed.
- R. Moncayo, A. Kroiss, M. O. F. Karakolcu, M. Starzinger, K. Kapelari, H. Talasz and H. Moncayo, BMC Endocr. Disord., 2008, 8, 2 CrossRef PubMed.
- A. Gottas, A. Ripel, F. Boix, V. Vindenes, J. Mørland and E. L. Oiestad, J. Pharmacol. Toxicol. Methods, 2015, 74, 75–79 CrossRef CAS PubMed.
- M. R. Moghadam, S. Dadfarnia, A. M. H. Shabani and P. Shahbazikhah, Anal. Biochem., 2011, 410, 289–295 CrossRef CAS PubMed.
- L. Li, H. Liu, Y. Shen, J. Zhang and J.-J. Zhu, Anal. Chem., 2011, 83, 661–665 CrossRef CAS PubMed.
- H.-T. Chang and E. S. Yeung, Anal. Chem., 1995, 67, 1079–1083 CrossRef CAS PubMed.
- Y. Leng, K. Xie, L. Ye, G. Li, Z. Lu and J. He, Talanta, 2015, 139, 89–95 CrossRef CAS PubMed.
- K. Jackowska and P. Krysinski, Anal. Bioanal. Chem., 2013, 405, 3753–3771 CrossRef CAS PubMed.
- X. Zheng, Y. Guo, J. Zheng, X. Zhou, Q. Li and R. Lin, Sens. Actuators, B, 2015, 213, 188–194 CrossRef CAS.
- N. Yusoff, A. Pandikumar, R. Ramaraj, H. N. Lim and N. M. Huang, Microchim. Acta, 2015, 182, 2091–2114 CrossRef CAS.
- J. Njagi, M. M. Chernov, J. C. Leiter and S. Andreescu, Anal. Chem., 2010, 82, 989–996 CrossRef CAS PubMed.
- X. Liu, H. Jiang, J. Lei and H. Ju, Anal. Chem., 2007, 79, 8055–8060 CrossRef CAS PubMed.
- V. Vinoth, J. J. Wu and A. M. Asiri, Sens. Actuators, B, 2015, 210, 731–741 CrossRef CAS.
- M. Zhang, K. Gong, H. Zhang and L. Mao, Biosens. Bioelectron., 2005, 20, 1270–1276 CrossRef CAS PubMed.
- Z. Wang, J. Liu, Q. Liang, Y. Wang and G. Luo, Analyst, 2002, 127, 653–658 RSC.
- N. G. Tsierkezos, S. H. Othman, U. Ritter, L. Hafermann, A. Knauer, J. M. Köhler, C. Downing and E. K. McCarthy, Sens. Actuators, B, 2016, 231, 218–229 CrossRef CAS.
- S. Ramakrishnan, K. R. Pradeep, A. Raghul, R. Senthilkumar, M. Rangarajan and N. K. Kothurkar, Anal. Methods, 2015, 7, 779–786 RSC.
- B. Zhang, D. Huang, X. Xu, G. Alemu, Y. Zhang, F. Zhan, Y. Shen and M. Wang, Electrochim. Acta, 2013, 91, 261–266 CrossRef CAS.
- Y.-R. Kim, S. Bong, Y.-J. Kang, Y. Yang, R. K. Mahajan, J. S. Kim and H. Kim, Biosens. Bioelectron., 2010, 25, 2366–2369 CrossRef CAS PubMed.
- Z.-H. Shenga, X.-Q. Zhenga, J.-Y. Xua, W.-J. Baoa, F.-B. Wanga and X.-H. Xia, Biosens. Bioelectron., 2012, 34, 125–131 CrossRef PubMed.
- X. Wang, M. Wu, W. Tang, Y. Zhu, L. Wang, Q. Wang, P. He and Y. Fang, J. Electroanal. Chem., 2013, 695, 10–16 CrossRef CAS.
- J. Yan, S. Liu, Z. Zhang, G. He, P. Zhou, H. Liang, L. Tian, X. Zhou and H. Jiang, Colloids Surf., B, 2013, 111, 392–397 CrossRef CAS PubMed.
- C.-L. Suna, H.-H. Lee, J.-M. Yang and C.-C. Wu, Biosens. Bioelectron., 2011, 26, 3450–3455 CrossRef PubMed.
- F. Li, J. Chai, H. Yang, D. Han and L. Niu, Talanta, 2010, 81, 1063–1068 CrossRef CAS PubMed.
- J. Li, J. Yang, Z. Yang, Y. Li, S. Yu, Q. Xu and X. Hu, Anal. Methods, 2012, 4, 1725–17289 RSC.
- A. Pandikumar, G. T. S How, T. P. See, F. S. Omar, S. Jayabal, K. Z. Kamali, N. Yusoff, A. Jamil, R. Ramaraj, S. A John, H. N. Lim and N. M. Huang, RSC Adv., 2014, 4, 63296–63323 RSC.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- S. Biswas, D. Chakraborty, R. Das, R. Bandyopadhyay and P. Pramanik, Anal. Chim. Acta, 2015, 890, 98–107 CrossRef CAS PubMed.
- S. Biswas, R. Das, D. Chakraborty, R. Bandhyopadhyay and P. Pramanik, Electroanalysis, 2015, 27, 1253–1261 CrossRef CAS.
- C. Gao, S. Moya, H. Lichtenfeld, A. Casoli, H. Fiedler, E. Donath and H. Möhwald, Macromol. Mater. Eng., 2001, 286, 355–361 CrossRef CAS.
- C. R. Raj and T. Ohsaka, J. Electroanal. Chem., 2001, 496, 44–49 CrossRef CAS.
- M. Zhang, K. Gong, H. Zhang and L. Mao, Biosens. Bioelectron., 2005, 20, 1270–1276 CrossRef CAS PubMed.
- C. Xiao, X. Chu, Y. Yang, X. Li, X. Zhang and J. Chen, Biosens. Bioelectron., 2011, 26, 2934–2939 CrossRef CAS PubMed.
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed. Engl., 2008, 47, 3696–3717 CrossRef CAS PubMed.
- Z.-H. Sheng, X.-Q. Zheng, J.-Y. Xu, W.-J. Bao, F.-B. Wang and X.-H. Xia, Biosens. Bioelectron., 2012, 34, 125–131 CrossRef CAS PubMed.
- Y. Wang, X. Zhang, Z. Meng and Y. Cai, ACS Appl. Mater. Interfaces, 2012, 4, 286–295 Search PubMed.
- H. Y. Yue, S. Huang, J. Chang, C. Heo, F. Yao, S. Adhikari, F. Gunes, L. C. Liu, T. H. Lee, E. S. Oh, B. Li, J. J. Zhang, T. Q. Huy, N. V. Luan and Y. H. Lee, ACS Nano, 2014, 8, 1639–1646 CrossRef CAS PubMed.
- J. Jiang and X. Du, Nanoscale, 2014, 6, 11303–11309 RSC.
- B. G. Dursun, Electroanalysis, 2010, 22, 1106–1114 CrossRef.
- S. Thiagarajan and S.-M. Chen, Talanta, 2007, 74, 212–222 CrossRef CAS PubMed.
- G. T. S. How, A. Pandikumar, H. N. Ming and L. H. Ngee, Sci. Rep., 2014, 4, 5044 CAS.
- P.-S. Teo, P. Alagarsamy, N.-M. Huang, H.-N. Lim and S. Yusran, Sensors, 2014, 14, 15227–15243 CrossRef PubMed.
- R. Nurzulaikha, H. N. Lim, I. Harrison, S. S. Lim, A. Pandikumar, N. M. Huang, S. P. Lim, G. S. H. Thien, N. Yusoff and I. Ibrahim, Sensing and Bio-Sensing Research, 2015, 5, 42–49 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: UV-VIS, Table S1 and real sample analysis Tables S3 and S4 and Fig. S3–S6. See DOI: 10.1039/c6ra16774h |
| This journal is © The Royal Society of Chemistry 2016 |