
Synthesis, mechanism and ethylene polymerization catalysis of Ge(IV), Sn(II) and Zr(IV) complexes derived from substituted β-diketiminates†
Xia Xiaoa,
Xiaomin Haoa,
Jianliang Baia,
Jianbin Chaob,
Wei Caob and
Xia Chen*a
aSchool of Chemistry and Chemical Engineering, Shanxi University, Taiyuan, 030006, China. E-mail: chenxia@sxu.edu.cn
bScientific Instrument Center, Shanxi University, Taiyuan, 030006, China
First published on 10th June 2016
Abstract
Insertion of LiCH(SiMe3)2 into the CN bond of the appropriate nitriles RCN afforded dimeric β-diketiminato lithium complexes [Li{N(SiMe3)C(R)C(H)C(R′)N(SiMe3)}]2 (1a, R = R′ = NMe2; 1b, R = But, R′ = o-C5H4N). Lithium salt 1a was used as a precursor to react with SnCl2 and GeCl4, respectively, and complexes [ClSn{N(SiMe3)C(NMe2)C(H)C(NMe2)N(SiMe3)}] (2) and [Cl2Ge{NC(NMe2)C(H)C(NMe2)N(GeCl3)}] (3) were obtained in good yields. By β-diketiminato lithium 1b ligand transfer, the cyclo-1,3-diazasilane heterocyclic complex  (4) was prepared using 1b and ZrCl4. Unexpectedly, the chelating β-diketiminato backbone in 3 acts as a dianionic ligand, whereas in 4, the ligand acts as a neutral cyclo-1,3-diazasilane heterocyclic ring to coordinate with the Zr center. The X-ray structures of 2–4 are presented, and reaction pathways for each complex are proposed. Upon activation with methylaluminoxane, complex 4 exhibits good activity for ethylene polymerization.
(4) was prepared using 1b and ZrCl4. Unexpectedly, the chelating β-diketiminato backbone in 3 acts as a dianionic ligand, whereas in 4, the ligand acts as a neutral cyclo-1,3-diazasilane heterocyclic ring to coordinate with the Zr center. The X-ray structures of 2–4 are presented, and reaction pathways for each complex are proposed. Upon activation with methylaluminoxane, complex 4 exhibits good activity for ethylene polymerization.
Introduction
Metal complexes supported by monoanionic β-diketiminato ligands have been widely explored1 mainly due to the modification in steric and electronic demands.2 The steric bulk of β-diketiminato ligands can be easily tuned by substituents on the nitrogen atoms or on the carbon framework. However, the variation of substituents attached to the carbon backbone of the ligands is rather limited.3 Thus, modification of the ligand skeleton can provide an opportunity to change their steric and electronic properties by coordination chemistry. In our former publications, we described the synthesis and structures of the β-diketiminato lithium compounds, in which the β-diketiminato ligands were prepared via insertion of various nitriles into the Li–C bond of LiCH(SiMe3)2.4–6 These β-diketiminato lithium compounds were ligand-transfer reagents when reacted with metal halides.7,8In this paper, we report the insertion products [Li{N(SiMe3)C(R)C(H)C(R′)N(SiMe3)}]2 (1a, R = R′ = NMe2; 1b, R = But, R′ = o-C5H4N) via LiCH(SiMe3)2 with nitriles,4,5 were used as ligand transfer reagents to react with metal chlorides. The precursor complexes 1a reacted with SnCl2 and GeCl4, respectively, to provide [ClSn{N(SiMe3)C(NMe2)C(H)C(NMe2)N(SiMe3)}] (2) and [Cl2Ge{NC(NMe2)C(H)C(NMe2)N(GeCl3)}] (3). The reaction of 1b with ZrCl4 afforded a cyclo-1,3-diazasilane Zr complex  (4). Interestingly, complex 3 contains a novel dianionic β-diketiminato ligand and 4 has a neutral cyclo-1,3-diazasilane heterocyclic ring, which is a new type of stable silicon compound. Complex 4 can initiate ethylene polymerization. Herein, the detailed investigations and discussions are described.
(4). Interestingly, complex 3 contains a novel dianionic β-diketiminato ligand and 4 has a neutral cyclo-1,3-diazasilane heterocyclic ring, which is a new type of stable silicon compound. Complex 4 can initiate ethylene polymerization. Herein, the detailed investigations and discussions are described.
Results and discussion
Synthesis and characterization of complexes 2 and 3
The β-diketiminato lithium salt (1a) was synthesized following the procedure reported in our previous publication.4 The reaction mechanism involves two consecutive nucleophilic attacks on a nitrile molecule, and each one is followed by a 1,3-shift of a trimethylsilyl group. Reaction of SnCl2 and GeCl4 with an equivalent of 1a in diethyl ether at −78 °C afforded the corresponding complex 2 (86%) as a white solid and 3 (65%) as a red solid, respectively. Complexes 2 and 3 are soluble in THF and Et2O but show poor solubility in hexanes (Scheme 1).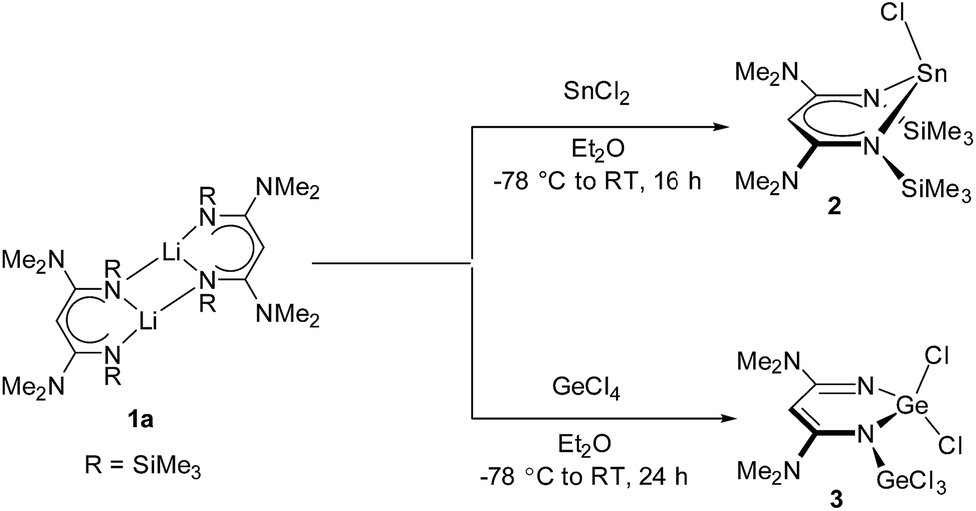 | ||
| Scheme 1 Synthetic pathway of complexes 2 and 3. | ||
We noticed that the reaction of 1a with GeCl4 afforded an unanticipated β-diketiminate 3, in which the end groups at the nitrogen atoms are not trimethylsilyl substituents. It was suggested that the silyl group is quite labile in the presence of Lewis acid GeCl4. Scheme 2 depicts a possible mechanism for the formation of 3. We hypothesized two steps to the formation of 3 from reactant 1a: (i) the Li compound 1a reacted with GeCl4 to form the corresponding β-diketiminate 3′ via a metathesis reaction; (ii) in the presence of excess GeCl4, an intermolecular reaction between 3′ and GeCl4 yields 3 via elimination of 2 equiv. Me3SiCl due to the Si–N bond being weaker than the Ge–N bond. For the elimination reaction of the chlorotrimethylsilane, it has been reported that the reaction of lithium amidinate [Me3SiNC(Ph)N(CH2)3N(Me)SiMe3]Li(THF) with TiCl4(THF)2 gave titanium amidinato amide dichloride complex [η2,η1-Me3SiNC(Ph)N(CH2)3NMe]TiCl2 by the elimination of LiCl and Me3SiCl.9 Our group demonstrated that lithium silylquinolylamide reacted with (MeC5H4)TiCl3 to afford titanium(IV) complex [{8-(C9H6N)NC(NMe2)N}Ti(MeC5H4)Cl]2 via a metathesis reaction and the elimination of SiMe3Cl.10 Apparently, in the presence of a stronger Lewis acid, the formation of a new metal–amido bond was thermodynamically favored by elimination of SiMe3Cl.
 | ||
| Scheme 2 Possible mechanism for the formation of 3. | ||
Crystals of 2 and 3 suitable for X-ray diffraction study were grown from a concentrated diethyl ether solution at −30 °C. The structures of 2 and 3, along with selected bond lengths and angles, are depicted in Fig. 1 and 2, respectively; the crystal and structural refinement data are summarized in Table 2.
 | ||
| Fig. 1 ORTEP (30% probability) diagram of 2. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Sn(1)–N(1) 2.154(6), Sn(1)–N(2) 2.187(6), Sn(1)–Cl(1) 2.471(2), N(1)–C(1) 1.362(9), N(2)–C(3) 1.350(9), C(1)–C(2) 1.408(9), C(2)–C(3) 1.396(9), N(1)–Sn(1)–N(2) 87.5(2), N(1)–Sn(1)–Cl(1) 95.51(17), N(2)–Sn(1)–Cl(1) 93.24(14). | ||
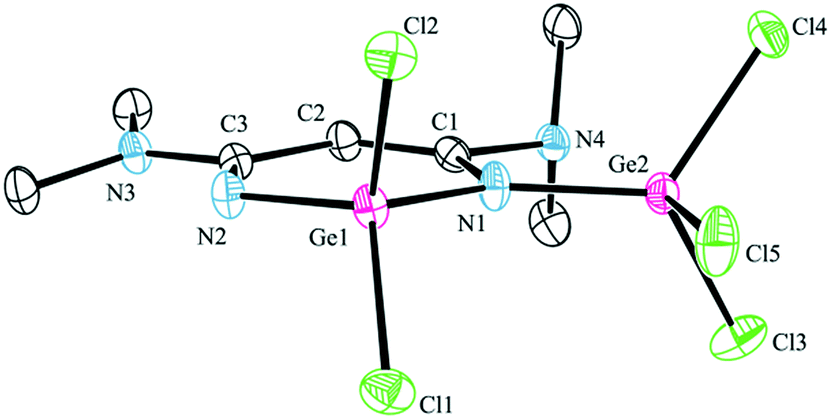 | ||
| Fig. 2 ORTEP (30% probability) diagram of 3. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Ge(1)–N(2) 1.765(3), Ge(1)–N(1) 1.844(3), Ge(2)–N(1) 1.838(3), N(1)–C(1) 1.371(5), N(2)–C(3) 1.297(5), C(1)–C(2) 1.329(6), C(2)–C(3) 1.472(5), N(2)–Ge(1)–N(1) 106.03(15), N(2)–Ge(1)–Cl(1) 112.23(12), N(1)–Ge(1)–Cl(1) 111.77(11), N(2)–Ge(1)–Cl(2) 112.81(12), N(1)–Ge(1)–Cl(2) 108.78(12), Cl(1)–Ge(1)–Cl(2) 105.30(5). | ||
In complex 2, the backbone of the chelating ligand (N1C1C2C3N2) is essentially planar (mean deviation: 0.0139 Å) and the tin atom is out of the plane (1.0601 Å); whereas the skeletal atoms including Ge (N1C1C2C3N2Ge six-membered ring) in complex 3, are almost coplanar with a mean deviation of 0.0282 Å. For β-diketiminato N1C1C2C3N2 moieties: in 2, the distances of C1–C2 and C2–C3 [1.408(9) and 1.396(9) Å] are rather the same, and the distances of N1–C1 and N2–C3 [1.362(9) and 1.350(9) Å] are between the bond lengths of C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds, indicating that a significant π-electron delocalization present as a η5 anion; in 3, the distances of C1–C2 and C2–C3 are 1.329(6) and 1.472(5) Å, and the distances of N(1)–C(1) and N(2)–C(3) are 1.371(5) and 1.297(5), which is consistent with localized β-diketiminate bonding over these atoms.
N bonds, indicating that a significant π-electron delocalization present as a η5 anion; in 3, the distances of C1–C2 and C2–C3 are 1.329(6) and 1.472(5) Å, and the distances of N(1)–C(1) and N(2)–C(3) are 1.371(5) and 1.297(5), which is consistent with localized β-diketiminate bonding over these atoms.
Analogues of 2 are reported in the following compounds: [HC(CMeNAr)2]SnCl (Ar = 2,6-i-Pr2C6H3),11 [HC{CPhN(SiMe3)}2]SnCl,12 [(Mes)2DAP]SnCl (where [(Mes)2DAP] = 2,4-dimethyl-N,1-N′-bis(2,4,6-trimethylphenyl)-1,5-diazapentadienyl),13 [{N(C6H3Pri2-2,6)C(H)}2CPh]SnCl,14 which were obtained from SnCl2 and the appropriate Li β-diketiminate. The fragment ClSnNN in 2 is similar to those analogues and the Sn–N bond lengths [2.154(6) and 2.187(6) Å] are in the normal range (2.121–2.397 Å).15
Complex 3 consists of two germanium atoms, both of them are in a distorted tetrahedral environment. It was worth noting that the distances of Ge(IV) to the amido nitrogen atom N1 [Ge(1)–N(1) = 1.844(3) and Ge(2)–N(1) = 1.838(3) Å] are longer than the distance of germanium to the imino nitrogen atom [Ge(1)–N(2) = 1.765(3) Å]; however, both of them are shorter than those of the corresponding Ge(II)–Nav in [CH{(CMe)(2,6-i-Pr2C6H3N)}2]GeCl (1.993 Å)16 or [CH{(CMe)(2,4,6-Me3C6H2N)}2]GeCl (1.993 Å).17
Synthesis and catalytic behavior of complex 4
The aza-allyl lithium starting material was obtained by the insertion of ButCN into LiCH(SiMe3)2.8,18–20 The synthetic procedure for zirconium complex 4 is illustrated in Scheme 3. The β-diketiminato lithium [Li{N(R)C(But)C(H)C(o-C5H4N)N(R)}]2 (R = SiMe3) (1b) is prepared from treatment of aza-allyl lithium with o-PyCN in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in diethyl ether.5
:1 molar ratio in diethyl ether.5
 | ||
| Scheme 3 Synthetic pathway for complex 4. | ||
Treatment of lithium precursor 1b (ref. 5) with an equimolar amount of ZrCl4 in toluene at −78 °C to room temperature for 24 h unexpectedly yielded the cyclo-1,3-diazasilane product 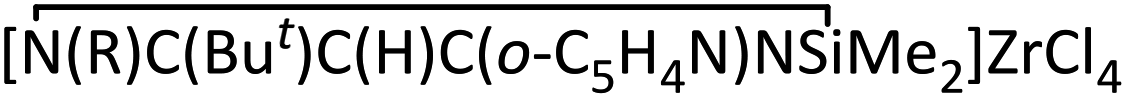 (4) instead of the β-diketiminato zirconium. Zr compound 4 was isolated in good yield (59%) as an orange solid by filtration of the resulting mixture to remove insoluble residue followed by recrystallization from toluene.
(4) instead of the β-diketiminato zirconium. Zr compound 4 was isolated in good yield (59%) as an orange solid by filtration of the resulting mixture to remove insoluble residue followed by recrystallization from toluene.
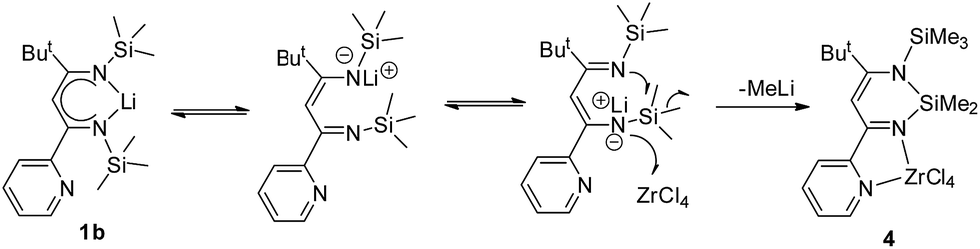
The formation of the silicon heterocyclic complex 4 involved Lewis-acid assisted cleavage of a Si–C bond and the formation of a new Si–N bond. The similar reaction was also reported by Leung and co-workers, in which the reaction of 2,3-pyrazyl-linked bis(1-aza-allyl) dilithium complex [Li2-{{N(SiMe3)C(But)C(H)}2C4H2N2-2,3}(THF)2]2 with CdCl2 afforded the silicon cyclized product  .21 The Lewis-acid assisted Si–C bond cleavage reaction was also applied in a methyl/chlorine exchange reaction triggered by the action of GaCl3.22 Herein, the unexpected formation of 4 in the reaction of 1b with ZrCl4 is presumably that of an intermolecular interaction of the weak acceptor, zirconium, with the anionic imido-nitrogen leading to the cleavage of an Si–C bond and the formation of a new Si–N bond with concomitant elimination of MeLi, which is a possible pathway to form the cyclized compound 4 (Scheme 4).
.21 The Lewis-acid assisted Si–C bond cleavage reaction was also applied in a methyl/chlorine exchange reaction triggered by the action of GaCl3.22 Herein, the unexpected formation of 4 in the reaction of 1b with ZrCl4 is presumably that of an intermolecular interaction of the weak acceptor, zirconium, with the anionic imido-nitrogen leading to the cleavage of an Si–C bond and the formation of a new Si–N bond with concomitant elimination of MeLi, which is a possible pathway to form the cyclized compound 4 (Scheme 4).
 | ||
| Scheme 4 Plausible mechanism for the formation of 4. | ||
The 1H NMR spectrum of 4 shows that the silylic CH3 protons are diastereotopic, appearing as two singlets at δ 0.05 and 0.91 ppm. As expected, the proton signals for SiMe2 are more deshielded than SiMe3. Likewise, the 13C NMR spectrum of 2 also indicates that the carbon chemical shift of SiMe2 (δ 3.81) is substantially more deshielded than SiMe3 (δ 3.30).
Single crystals of 4 suitable for X-ray diffraction were obtained at −30 °C after two weeks. In compound 2, the neutral ligand is coordinated to the metal in a κ2-fashion with the two nitrogen atoms from the cyclo-1,3-diazasilane and pyridyl (Fig. 3). The zirconium atom exhibits a distorted octahedral geometry furnished with two N atoms of ligand and four chlorine atoms. Both dative Zr1–N1 and Zr1–N2 bond distances of 2.328(4) and 2.283(4) Å, respectively, are comparable to those dative Zr–N interactions in bis{3-(2-pyridyl)-1-azaallyl}zirconium system [Zr{N(SiMe3)C(But)C(H)(C5H4N-2)}2Cl2] (2.322(3) Å) and [Zr{N(SiMe3)C(Ph)C(SiMe3)(C5H4N-2)}2Cl2] (2.354(3) Å),23 and pyridyl imido zirconium complexes [Zr{(2-C5H4N)C-(CH3){CH2NSi(CH3)2tBu}2(NDIPP)(Py)}] (Zr–NPy = 2.315(2) Å) and [Zr{(2-C5H4N)C-(CH3){CH2NSi(CH3)2tBu}2(NMesN2N(Ph))}] (Zr–NPy = 2.338(2) Å).24 These dative bond distances are significantly longer than the Zr–N covalent bond in the pyridyl imido zirconium complexes (Zr–Nimido = 2.096(2) and 2.104(2) Å),23 and amido complex (Zr–Namido = 2.141(2)–2.231(3) Å).22 For the cyclo-1,3-diazasilane skeleton, the bond lengths of N(2)–C(6) [1.309(6) Å] and C(7)–C(8) [1.362(6) Å] are significantly shorter than N(3)–C(8) [1.407(6) Å] and C(6)–C(7) [1.441(6) Å], indicating the presence of localized CC and CN double bonds; a similar phenomenon is also found in  .21
.21
 | ||
| Fig. 3 ORTEP (30% probability) diagram of 4. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Zr(1)–N(1) 2.328(4), Zr(1)–N(2) 2.283(4), N(1)–C(4) 1.355(6), N(2)–C(6) 1.309(6), C(4)–C(6) 1.494(6), C(6)–C(7) 1.441(6), C(7)–C(8) 1.362(6), N(3)–C(8) 1.407(6), Si(1)–N(2) 1.810(4), Si(1)–N(3) 1.756(4), N(2)–Zr(1)–N(1) 70.25(13), Cl(1)–Zr(1)–Cl(4) 92.05(5), N(2)–Zr(1)–Cl(1) 160.33(10), N(1)–Zr(1)–Cl(3) 168.24(9), Cl(2)–Zr(1)–Cl(4) 167.43(5), N(1)–Zr(1)–Cl(1) 90.33(11), N(2)–Zr(1)–Cl(3) 98.00(11). | ||
Compound 4 was investigated for its catalytic behavior of ethylene polymerization with methylaluminoxane (MAO) as a co-catalyst under atmospheric pressure. The results of the optimization conditions are summarized in Table 1.
| Entry | Al/Zr | Cat. (μmol) | T (°C) | Time (h) | Yield (mg PE) | Activityb | Tmh (°C) | 105Mwi | Mw/Mn |
|---|---|---|---|---|---|---|---|---|---|
| a Conditions: complex, 5 μmol; solvent, toluene; Vtotal = 100 mL; ethylene pressure, 1 atm.b Activity in units of 104 g of PE (mol of Zr)−1 h−1 atm−1.c The complex is ZrCl4. 5 atm of ethylene pressure.25d The complex is ZrCl4(THF)2.25e The complex is ZrCl4(Et2O)2.25f The complex is Cp2Zr2Cl2.26g The complex is dimethyl-β-IAM–ZrCl3. 6.8 atm of ethylene pressure.27h Determined by DSC.i Determined by GPC. | |||||||||
| 1 | 200 | 5 | 25 | 0.5 | 43.1 | 1.72 | 138.2 | 8.78 | 3.17 |
| 2 | 500 | 5 | 25 | 0.5 | 64.5 | 2.58 | 138.0 | 10.5 | 3.6 |
| 3 | 1000 | 5 | 25 | 0.5 | 38.2 | 1.52 | 136.2 | 9.28 | 5.7 |
| 4 | 500 | 5 | 25 | 1.0 | 108 | 2.16 | 138.8 | 11.5 | 5.7 |
| 5 | 500 | 5 | 50 | 0.5 | 560 | 22.4 | 137.8 | 12.4 | 6.7 |
| 6c | 500 | 10 | 50 | 1.0 | 950 | 0.37 | n.d. | n.d. | |
| 7d | 500 | 10 | 50 | 0.5 | 1100 | 22.0 | 140.6 | 12.3 | 5.9 |
| 8e | 500 | 10 | 50 | 0.5 | 1200 | 24.0 | 142.8 | 13.8 | 5.9 |
| 9f | 500 | 10 | 50 | 1.0 | 4500 | 45.8 | 137.5 | n.d. | |
| 10g | 1000 | 2.0 | 25 | 1.0 | 560 | 4.51 | 138.1 | n.d. | |
At the temperature of 25 °C, with an increase of the molar ratios of Al to Zr from 200 to 1000 (entries 1–3), the catalytic ability of 4 achieved the maximum activity of 2.58 × 104 g of PE (mol of Zr)−1 h−1 atm−1 at a Al/Zr molar ratio of 500 (entry 2) and then decreased to a lower value of 1.52 × 104 g of PE (mol of Zr)−1 h−1 atm−1 at a Al/Zr molar ratio of 1000 (entry 3). At different times (0.5 to 1 h, entries 2 and 4), the yield increased with the increase of polymerization time. This behavior indicated that the catalyst had a catalytic lifetime of at least 1 h. The reaction temperature also influenced the catalytic performance significantly, the activities were increased from 2.58 × 104 to 22.4 × 104 g of PE (mol of Zr)−1 h−1 atm−1 at 50 °C (entry 5). This result showed that raising the temperature could accelerate the formation of the active species.
The activity of 4 (entry 5) is about a hundred times higher than ZrCl4 (entry 6), but comparable to those complexes of ZrCl4 incorporating donor reagents such as ZrCl4(THF)2 (entry 7) or ZrCl4(Et2O)2 (entry 8).25 As can be noted, 4/MAO is only two times less active than metallocene pre-catalyst Cp2ZrCl2 (entries 5 and 9) under similar conditions;26 therefore, this catalyst mixture can be described as high activity according to the Gibson classification.27a Comparison with β-diketiminate Zr complexes, such as dimethyl-β-iminoaminate (IAM)–ZrCl3 [4.51 × 104 g of PE (mol of Zr)−1 h−1 atm−1, entry 10]27b and [(2-Pyr)C(H)C(But)N(SiMe3)]ZrCl3 [7.50 × 104 g of PE (mol of Zr)−1 h−1 atm−1],27a the activity of complex 4 is an order of magnitude higher than those of β-diketiminates. The PEs produced by 4/MAO have high melting temperatures in the range of 136.2–138.8 °C, typical for high-density polyethylene, which are lower than those of the polyethylenes obtained by ZrCl4(THF)2 and ZrCl4(Et2O)2 (entries 7 and 8),25 and comparable to those of the polyethylenes formed with zirconocenes (entries 9 and 10). The GPC analyses show a wide molecular weight distribution because of the formation of more than one active species in the reaction of 4 with MAO.
Conclusion
In summary, the unusual X-ray characterized crystalline Sn(II), Ge(IV) and Zr(IV) complexes have been prepared via a salt metathesis and elimination reaction. Their novelty lies in the phenomenon that a β-diketiminato ligand in each complex is very different: (i) for the Sn(II) compound, the β-diketiminato ligand is monoanionic in η5-fashion; (ii) for the Ge(IV) compound, the β-diketiminato ligand is dianionic with localized bonding over these atoms of backbone. And (iii) for the Zr(IV) compound, the ligand is a neutral cyclo-1,3-diazasilane heterocyclic ring, which represents a new type of silicon-bridged compound. The possible mechanistic pathways for the formation of complexes 3 and 4 have been proposed. The catalytic behavior of complex 4 was investigated in the presence of MAO as a co-catalyst, and the results showed that pre-catalyst 4 has high activity for ethylene polymerization up to 22.4 × 104 g of PE (mol of Zr)−1 h−1.Experimental section
Materials and procedures
All manipulations were carried out under an inert nitrogen atmosphere using standard Schlenk techniques. Solvents were dried with sodium, distilled from sodium/potassium alloy (toluene), sodium/benzophenone (diethyl ether) and stored over molecular sieves (4 Å). Deuterated C6D6 was dried over activated molecular sieves (4 Å). All solvents were degassed prior to use. Chemicals were purified by distillation before use. The β-diketiminato lithium [Li{N(SiMe3)C(NMe2)C(H)C(NMe2)N(SiMe3)}]2 (1a)4 and [Li{N(SiMe3)C(But)C(H)C(o-C5H4N)N(SiMe3)}]2 (1b)5 were prepared according to the reported procedures. 1H and 13C NMR spectra were recorded on a Bruker DRX-300. Elemental analyses were carried out with a Vario EL-III instrument. The molecular weights of polyethylene were measured by a PL-GPC at 150 °C using 1,2,4-trichlorobenzene as the eluent and calibrated by polystyrene standards. Transition melting temperatures (Tm) were measured on a Perkin-Elmer DSC-7 differential scanning calorimeter, measured upon reheating the polymer sample to 180 °C at a heating rate of 20 °C min−1. (4). ZrCl4 (0.71 g, 3.03 mmol) was added to a solution of 1b (1.12 g, 3.02 mmol) in toluene at −78 °C. The mixture was warmed to room temperature and stirred for a further 24 h. Then, the solution was filtered off and concentrated. The product was isolated by crystallization from toluene for 7 d at −30 °C to give yellow crystals 4. Yield: 1.01 g (59%). 1H NMR (C6D6, 300 MHz): δ 9.58 (s, 1H, o-py), 8.17 (d, 1H, J = 8.7 Hz, o-py), 7.93 (s, 1H, o-py), 7.09 (s, 1H, o-py), 2.34 (s, 1H, CH), 1.47 (s, 9H, C(CH3)3), 0.91 (s, 6H, Si(CH3)2), 0.05 (s, 9H, Si(CH3)3). 13C{1H} NMR (C6D6, 75 MHz): δ 181.1, 161.5, 149.7, 138.7, 122.8, 96.8, 99.7, 98.6, 44.5, 29.4, 3.8, 3.3. Anal. calcd for C17H29Cl4N3Si2Zr: C, 36.16; H, 5.18; N, 7.44. Found: C, 36.08; H, 5.08; N, 7.40.
(4). ZrCl4 (0.71 g, 3.03 mmol) was added to a solution of 1b (1.12 g, 3.02 mmol) in toluene at −78 °C. The mixture was warmed to room temperature and stirred for a further 24 h. Then, the solution was filtered off and concentrated. The product was isolated by crystallization from toluene for 7 d at −30 °C to give yellow crystals 4. Yield: 1.01 g (59%). 1H NMR (C6D6, 300 MHz): δ 9.58 (s, 1H, o-py), 8.17 (d, 1H, J = 8.7 Hz, o-py), 7.93 (s, 1H, o-py), 7.09 (s, 1H, o-py), 2.34 (s, 1H, CH), 1.47 (s, 9H, C(CH3)3), 0.91 (s, 6H, Si(CH3)2), 0.05 (s, 9H, Si(CH3)3). 13C{1H} NMR (C6D6, 75 MHz): δ 181.1, 161.5, 149.7, 138.7, 122.8, 96.8, 99.7, 98.6, 44.5, 29.4, 3.8, 3.3. Anal. calcd for C17H29Cl4N3Si2Zr: C, 36.16; H, 5.18; N, 7.44. Found: C, 36.08; H, 5.08; N, 7.40.General procedure for ethylene polymerization
X-ray crystallography
Data collection of 2, 3 and 4 was performed with Mo-Kα radiation (λ = 0.71073 Å) on a Bruker Smart Apex CCD diffractometer. The structures were solved by direct methods (SHELXS-97)28 and refined against F2 by full-matrix least squares using SHELXL-97.29 All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were placed in calculated positions. Crystal data and experimental details of the structure determinations are listed in Table 2.| Complex | 2 | 3 | 4 |
|---|---|---|---|
| Formula | C13H31ClN4Si2Sn | C7H13Cl5Ge2N4 | C17H29Cl4N3Si2Zr |
| Mw (g mol−1) | 455.75 | 475.64 | 564.63 |
| T (K) | 183(2) K | 456(2) K | 293(2) K |
| Crystal system | Monoclinic | Triclinic | Triclinic |
| Space group | P21/n | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| a (Å) | 11.7137(17) | 8.7293(16) | 10.139(2) |
| b (Å) | 15.426(2) | 9.6018(18) | 12.050(2) |
| c (Å) | 11.7657(17) | 10.0436(19) | 12.659(3) |
| α (°) | 90 | 91.651(2) | 118.34(3) |
| β (°) | 91.121(2) | 98.999(2) | 110.25(3) |
| γ (°) | 90 | 102.452(2) | 91.62(3) |
| V (Å3) | 2125.6(5) | 810.2(3) | 1241.9(4) |
| Z, Dcalcd (g cm−3) | 4, 1.424 | 2, 1.950 | 2, 1.510 |
| μ (mm−1) | 1.441 | 4.522 | 0.978 |
| F(000) | 936 | 464 | 576 |
| Reflections collected | 8605 | 3339 | 5135 |
| Independent (Rint) reflections | 3697(0.0427) | 2782(0.0166) | 4291(0.0255) |
| Goodness of fit on F2 | 1.015 | 1.010 | 1.048 |
| Final R indices [I > 2σ(I)] R1, wR2 | 0.0517, 0.1577 | 0.0326, 0.0780 | 0.0545, 0.1120 |
| R indices (all data) R1, wR2 | 0.0626, 0.1745 | 0.0402, 0.0803 | 0.0674, 0.1181 |
| Largest diff. peak and hole [e Å−3] | 0.580 and −1.024 | 0.580 and −1.024 | 1.064 and −0.411 |
Acknowledgements
We thank the National Natural Science Foundation of China (grant no. 21072120), Shanxi International Science and Technology Cooperation Program (2015081050) and Shanxi Patent Promotion Implementation Project (20161001).References
- (a) Y. C. Tsai, Coord. Chem. Rev., 2012, 256, 722 CrossRef CAS; (b) L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031 CrossRef CAS PubMed.
- P. H. M. Budzelaar, A. B. Van Oort and A. G. Orpen, Eur. J. Inorg. Chem., 1998, 1485 CrossRef CAS.
- S. Yokota, Y. Tachi, N. Nishiwaki, M. Ariga and S. Itoh, Inorg. Chem., 2001, 40, 5316 CrossRef CAS PubMed.
- X. Chen, C. X. Du, J. P. Guo, X. H. Wei and D. S. Liu, J. Organomet. Chem., 2002, 655, 89 CrossRef CAS.
- X. Chen, L. Wang, J. P. Guo, S. P. Huang and D. S. Liu, Mendeleev Commun., 2005, 4, 160 CrossRef.
- X. Chen, L. Wang, S. P. Huang and D. S. Liu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, o473 CrossRef CAS.
- X. Chen, L. L. Guan, M. S. Eisen, H. F. Li, H. B. Tong, L. P. Zhang and D. S. Liu, Eur. J. Inorg. Chem., 2009, 3488 CrossRef CAS.
- P. B. Hitchcock, M. F. Lappert and D. S. Liu, J. Chem. Soc., Chem. Commun., 1994, 2637 RSC.
- W. J. Meerendonk, K. Schröder, E. A. C. Brussee, A. Meetsma, B. Hessen and J. H. Teuben, Eur. J. Inorg. Chem., 2003, 427 CrossRef.
- P. Wang, H. Li, X. Xue and X. Chen, Dalton Trans., 2015, 44, 4718 RSC.
- Y. Ding, H. W. Roesky, M. Noltemeyer, H.-G. Schmidt and P. P. Power, Organometallics, 2001, 20, 1190 CrossRef CAS.
- P. B. Hitchcock, M. F. Lappert and J. R. Severn, Dalton Trans., 2004, 4193 RSC.
- A. E. Ayers, T. M. Klapötke and H. R. V. Dias, Inorg. Chem., 2001, 40, 1000 CrossRef CAS.
- D. J. Doyle, P. B. Hitchcock, M. F. Lappert and G. Li, J. Organomet. Chem., 2009, 694, 2611 CrossRef CAS.
- C. Cui, H. W. Roesky, H. Hao, H.-G. Schmidt and M. Noltemeyer, Angew. Chem., 2000, 112, 1888 (Angew. Chem., Int. Ed., 2000, 39, 1815) CrossRef.
- Y. Ding, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, Organometallics, 2001, 20, 1190 CrossRef CAS.
- A. E. Ayers, T. M. Klapötke and H. V. R. Dias, Inorg. Chem., 2001, 40, 1000 CrossRef CAS.
- P. B. Hitchcock, M. F. Lappert, W. P. Leung, D. S. Liu, T. C. W. Mak and Z. X. Wang, J. Chem. Soc., Dalton Trans., 1999, 1263 RSC.
- M. F. Lappert and D. S. Liu, J. Organomet. Chem., 1995, 500, 203 CrossRef CAS.
- P. B. Hitchcock, M. F. Lappert, M. Layh, D. S. Liu, R. Sablong and T. Shun, J. Chem. Soc., Dalton Trans., 2000, 14, 2301 RSC.
- W. P. Leung, W. Y. I. Queenie, T. W. Lam and T. C. W. Mak, Organometallics, 2004, 23, 1284 CrossRef CAS.
- D. Michalik, A. Schulz and A. Villinger, Inorg. Chem., 2008, 47, 11798 CrossRef CAS PubMed.
- B. J. Deelman, P. B. Hitchcock, M. F. Lappert, W. P. Leung, H. K. Lee and T. C. W. Mak, Organometallics, 1999, 18, 1444 CrossRef CAS.
- T. Gehrmann, J. L. Fillol, H. Wadepohl and L. H. Gade, Organometallics, 2012, 31, 4504 CrossRef CAS.
- A. Proto, C. Capacchione, O. Motta and F. D. Carlo, Macromolecules, 2003, 36, 5942 CrossRef CAS.
- (a) W. Kaminsky and R. Steiger, Polyhedron, 1988, 4, 2357 Search PubMed; (b) W. Kaminsky, Macromol. Chem. Phys., 1996, 197, 3907 CrossRef CAS.
- (a) V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283 CrossRef CAS PubMed; (b) X. Jin and B. M. Novak, Macromolecules, 2002, 33, 6205 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 467 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † CCDC 1450544–1450546. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra11097e |
| This journal is © The Royal Society of Chemistry 2016 |