
Comparative studies of the electronic structure and thermoelectric properties in orthorhombic and tetragonal BaCu2Se2 by first-principles calculations
Daifeng Zouab,
Hairong Zheng*b and
Jiangyu Li*c
aSchool of Physics and Electronic Science, Hunan University of Science and Technology, Xiangtan 411201, China
bShenzhen Key Laboratory of Nanobiomechanics, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, China. E-mail: hr.zheng@siat.ac.cn
cDepartment of Mechanical Engineering, University of Washington, Seattle, WA 98195-2600, USA. E-mail: jjli@u.washington.edu
First published on 8th June 2016
Abstract
The electronic structures of BaCu2Se2 in two phases, orthorhombic α-BaCu2Se2 and tetragonal β-BaCu2Se2, are investigated by using first-principles calculations. For the two phases, it is found that the bottom of the conduction band primarily comes from Ba 5d orbitals while the upper valence band consists of Cu 3d and Se 4p orbitals, and both of them exhibit direct band gaps. The calculated electronic structures reveal that α-BaCu2Se2 has a higher Seebeck coefficient while β-BaCu2Se2 possesses a larger electrical conductivity. The thermoelectric properties of p-type α- and β-BaCu2Se2 are calculated on the base of the semi-classical Boltzmann transport theory. It is observed that the thermoelectric performance of tetragonal β-BaCu2Se2 is superior to orthorhombic α-BaCu2Se2. The optimal doping concentrations have been estimated based on the predicted maximum power factors, and the temperature dependence of Seebeck coefficient of β-BaCu2Se2 is also estimated and compared with experimental data, with good agreements observed.
1. Introduction
Thermoelectric materials have become a promising research topic for the past several decades based on their ability to convert waste heat directly into useful electrical energy.1,2 The conversion efficiency of thermoelectric materials is characterized by the dimensionless figure of merit ZT = S2σT/κ, where S is the Seebeck coefficient, σ is the electrical conductivity, and κ is the thermal conductivity that generally consists of the electric (κe) and lattice (κl) contributions. A favorable thermoelectric ZT figure must simultaneously possess high Seebeck coefficient S, high electrical conductivity σ and low thermal conductivity κ. However, achieving all these requirements together in a particular material is challenging because these different parameters governing ZT are coupled and compete with each other. To enhance ZT, several successful concepts, such as solid-solution alloying and nanostructuring,3,4 have been developed over the last two decades to reduce the lattice thermal conductivity. Alternatively, one can also seek high thermoelectric performance in pristine materials with intrinsically low thermal conductivity by systematically optimizing their electronic properties.5,6Recently, the ternary copper chalcogenide BaCu2Se2 has been reported to exhibit excellent thermoelectric performance.7–9 This compound possesses high Seebeck coefficient S (390 μV K−1) and low thermal conductivity κ (1.5 W m−1 K−1) at room temperature,7,8 which may be a promising candidate for the next generation thermoelectric devices, though it has a moderate electrical conductivity (5.5 S cm−1) at low temperature. BaCu2Se2 is one of the family of related compounds BaCu2Ch2 (Ch = S, Se and Te), which consists two phases, namely, orthorhombic α-BaCu2Se2 and tetragonal β-BaCu2Se2.7 It was reported that the electrical conductivity of α-BaCu2Se2 can be improved by introducing Na doping while maintaining a relatively high Seebeck coefficient and low thermal conductivity, resulting in a ZT value of 1.0 at 773 K.7 The thermoelectric properties of BaCu2Se2 by potassium doping has been investigated by Zhang et al., and it was found that the figure of merit ZT can reach 0.32 at 800 K, mainly due to the one order of magnitude higher electrical conductivity improved by doping.8 Furthermore, the electronic and optical properties of BaCu2Se2 were measured, and electrical resistivity, Seebeck coefficient, and thermal conductivity were also presented.9 Additionally, the structure, physical and transport properties of related compounds, BaCu2S2 and BaCu2Te2, have been investigated experimentally.10–14
On the theoretical side, the electronic structures of BaCu2S2 and BaCu2Se2 are studied by density functional theory, and the band gaps and characteristics are analyzed.15,16 Combining with the results of the electronic and optical measurements, McGuire et al. investigated the electronic structures and transport properties of α-BaCu2Se2 and α-BaCu2Te2 using first-principles calculations.9 Additionally, the electronic structure and thermoelectric properties of β-BaCu2S2 were investigated using first-principles calculations by Jiao et al., and the feature of electronic structure and transport properties of β-BaCu2S2 were presented.17 However, few studies up to now have theoretically investigated the electronic structures and thermoelectric performance of BaCu2Se2. In order to further enhance the thermoelectric performance of BaCu2Se2, it is necessary to make a comparison of electronic structures and transport properties of orthorhombic α-BaCu2Se2 and tetragonal β-BaCu2Se2, and to predict their thermoelectric performance based on theoretical analysis. In this paper, we report electronic structures and transport properties of α- and β-BaCu2Se2 using first-principles calculations and semi-classical Boltzmann transport theory, and explore the interplay of their band structures and transport behaviors of the two phases. We then make a comparison of their thermoelectric performance and estimate the optimal doping concentrations of α-BaCu2Se2 and β-BaCu2Se2.
2. Computational method
The structural and electronic properties of α- and β-BaCu2Se2 are investigated using the first-principles as implemented in Vienna ab initio Simulation Package (VASP).18 The projector augment wave (PAW)19 pseudopotentials are used to describe the ionic potentials of the elements. The revised Perdew–Burke–Ernzerhof (revPBE)20 generalized gradient approximation (GGA) and Heyd–Scuseria–Ernzerhof (HSE) hybrid functional21 are employed as the exchange-correlation functional in band structure calculations. The cutoff energy of the plane-wave was both set at 400 eV. The energy convergence criterion were chosen to be 10−6 eV per unit cell, and the forces on all relaxed atoms were less than 0.03 eV Å−1. It is well known that density functional theory (DFT) often fail in the accurate description of atoms with strongly correlated electrons.22 Therefore, in this paper we have chosen to use the DFT plus U (DFT + U) method aiming at a more accurate description of the electronic properties of the Cu-based materials. In this method a Hubbard-like term (+U) is introduced in order to correctly describe the on-site Coulomb repulsion, leading to a more realistic localization of d-electrons on Cu atoms. The effective Coulomb repulsion parameter U was set to be 4 eV based on the published literature of Cu-based ternary semiconductors.23 The HSE screening parameter was set to a value of 0.2 Å−1.The Seebeck coefficient S and electronic conductivity over relaxation time σ/τ were obtained using the semi-classical Boltzmann theory in conjunction with rigid band and constant relaxation time approximations. All the calculations of transport properties were implemented in the BoltzTraP package24 that has successfully predicted the temperature and carrier concentration dependence of transport properties for some thermoelectric materials.23,25–27 The necessary crystal structures and eigen-energies for BoltzTraP calculation were obtained from revPBE + U results. In order to get reasonable transport properties, the Brillouin zones of the unit cells were represented by the Monkhorst–Pack special k-point scheme with 14 × 32 × 12 and 31 × 31 × 31 meshes for the orthorhombic α-BaCu2Se2 and tetragonal β-BaCu2Se2 phases, respectively. This provides well-converged transport quantities.
3. Results and discussion
3.1 Crystal structure
The primitive cell of α-BaCu2Se2 contains twenty atoms and has an orthorhombic structure with space group Pnma, as shown in Fig. 1(a). It is composed of a three dimensional Cu–Se network with Ba atoms residing in it.9 The tetragonal β-BaCu2Se2 is crystallized in the ThCr2Si2-type structure with I4/nmm space group. The conventional cell of the β-BaCu2Se2 is shown in Fig. 1(b), which has a unique crystal structure wherein Cu2Se2 layers are sandwiched by Ba sheets like a natural superlattice. The optimized lattice constants of α-BaCu2Se2 and β-BaCu2Se2 are listed in Table 1 together with experimental data.9,28 As we can see from Table 1, the calculated values of α-BaCu2Se2 and β-BaCu2Se2 overestimate the lattice constants within 1% compared with experimental values reported,9 and it is well known that GGA often overestimates lattice constants of solids.29 Meanwhile, it can be seen from Table 1 that they clearly demonstrate that the HSE functional systematically improves upon the revPBE lattice constants, and this is consistent with the previous report.15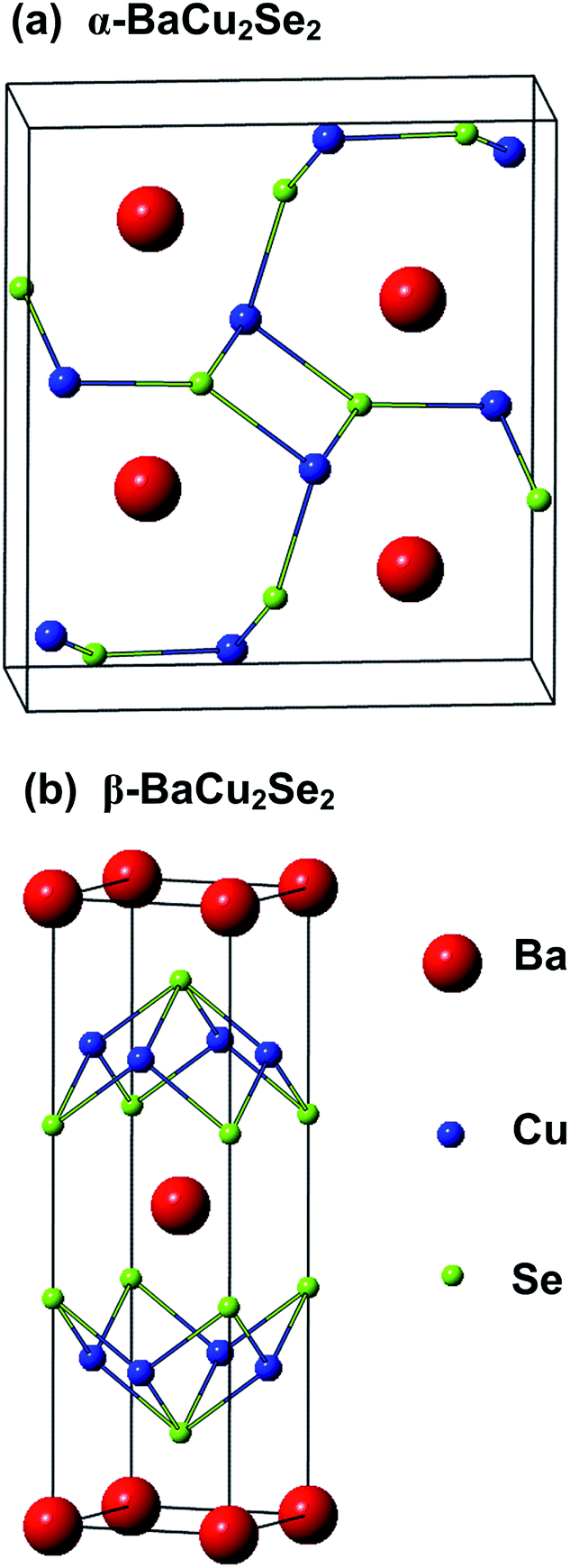 | ||
| Fig. 1 Crystal structure of (a) orthorhombic α-BaCu2Se2 and (b) tetragonal β-BaCu2Se2. | ||
3.2 Electronic structure
The calculated electronic band structures of α-BaCu2Se2 and β-BaCu2Se2 along the high symmetry directions are shown in Fig. 2 using both the revPBE + U and HSE approaches. It can be seen from Fig. 2 that both α-BaCu2Se2 and β-BaCu2Se2 shows direct band gap feature as reported for BaCu2S2,16 that is, the conduction band minimum (CBM) and the valence band maximum (VBM) are both located at Γ. The calculated energy gaps with revPBE + U are found to be 0.85 eV and 0.75 eV for α-BaCu2Se2 and β-BaCu2Se2, respectively, which are lower than these values calculated by the HSE method (1.38 eV for α-BaCu2Se2 and 0.98 eV for β-BaCu2Se2), and this is due to the problems associated with revPBE + U used in our calculations, which still cannot entirely describe the exchange-correlation effects of the localized Cu 3d electrons.25 The HSE result of α-BaCu2Se2 matches well with the value reported previously (1.33 eV).15 Meanwhile, we can see from Fig. 2 that the valence band edge structures in the revPBE + U and HSE methods are very similar. Based on semi-classic Boltzmann transport theory, thermoelectric properties of semiconductors are determined by the band edge features. Considering the computational cost, we choose the revPBE + U method for further electronic structure and transport calculations. In order to obtain more realistic thermoelectric properties, we correct this problem of band gap using a scissors operator by moving the conduction band upward to match the HSE gap in the transport properties calculations due to the HSE method gives a band gap close to experiment, as previously reported for some semiconductors.23,30 | ||
| Fig. 2 Calculated band structures of α-BaCu2Se2 and β-BaCu2Se2 using the revPBE + U and HSE methods. The Fermi levels are set to zero. | ||
Since experimental works to date have found that BaCu2Se2 tends to form p-type semiconductors,7–9 our discussion focus on the upper part of the valence band. From the upper part of the valence band near the Fermi level as shown in Fig. 2, we can see that the band dispersion of α-BaCu2Se2 at the Γ-Y and Γ-Z lines are more flat than the one of β-BaCu2Se2 at the Γ-M and Γ-Z lines. According to previous reports,23,25–27 the valence band near the Fermi level determines the transport properties of p-type thermoelectric materials. As the band dispersion varied from flat to steep, the effective mass becomes smaller, and it will increase the electrical conductivity. In order to quantitatively analyze this, we apply effective-mass calculations to understand the difference of α- and β-BaCu2Se2. The effective mass m* is defined in band theory as
 | (1) |
| Direction | α-BaCu2Se2 | β-BaCu2Se2 | ||
|---|---|---|---|---|
| Γ-Y | Γ-Z | Γ-M | Γ-Z | |
| mh (revPBE) | 3.1405 | 2.9646 | 0.2382 | 0.1621 |
| mh (HSE) | 2.8993 | 3.0966 | 0.2937 | 0.1747 |
In order to further understand the electronic structures of BaCu2Se2, we plot the total and projected density of states (DOS) of α-BaCu2Se2 and β-BaCu2Se2 in the energy interval between −7 eV and 5 eV in Fig. 3. For the two phases, the projected DOS indicates that the bottom of the conduction band primarily comes from Ba 5d orbitals, and the top of the valence band is mainly due to the hybridization of Cu 3d and Se 4p orbitals. Since BaCu2Se2 tends to form p-type semiconductor,7–9 our interest is in the upper valence band. The upper valence bands of BaCu2Se2 are mainly composed of Cu d and Se p orbitals, and contain no obvious contribution from Ba atoms. Now that the Ba 5d orbitals have a negligible contribution to valence band, it is expected that p-doping at the Ba-site will only change the carrier concentrations with little effect on the shape of the valence band maximum. In fact, some experimental works have been reported that the thermoelectric performance of BaCu2Se2 can be enhanced by doping of the alkali metals in the Ba sites.7,8
 | ||
| Fig. 3 Calculated total and projected density of states of (a) α-BaCu2Se2 and (b) β-BaCu2Se2 from the revPBE + U method. | ||
To further discuss the subtle changes of the electronic structures, the DOS of α-BaCu2Se2 and β-BaCu2Se2 near the band gap are shown in Fig. 4. It is well known that the slope of the DOS near the Fermi level can reflect the transport properties of thermoelectric materials,31,32 and a rapid change in DOS with energy is a good indicator of large Seebeck coefficient. As shown in Fig. 4, the slope of the α-BaCu2Se2 is steeper than β-BaCu2Se2 near the upper valence band, suggesting that α-BaCu2Se2 has larger Seebeck coefficient than β-BaCu2Se2.
 | ||
| Fig. 4 Total density of states of (a) α-BaCu2Se2 and (b) β-BaCu2Se2 near Fermi level in revPBE + U method. The valence-band maximum are set to zero. | ||
3.3 Thermoelectric properties
To make a comparison in the thermoelectric performance between the two phases of BaCu2Se2, transport properties of α- and β-BaCu2Se2 were calculated by solving Boltzmann transport equation. The electronic transport coefficients of p-type α-BaCu2Se2 and β-BaCu2Se2 as a function of number of holes per unit cell at 300 K and 800 K are shown in Fig. 5. As we can see from Fig. 5(a), the Seebeck coefficients of the two phases decrease with increased doping concentration. At the same doping level, the Seebeck coefficients decrease with increasing temperature. Furthermore, the Seebeck coefficients of α-BaCu2Se2 are larger than β-BaCu2Se2, which is consistent with the previous analysis of the DOS of α-BaCu2Se2 and β-BaCu2Se2 near the band gap. Here we will make a comparison of Seebeck coefficients among tetragonal BaCu2S2, BaCu2Se2 and BaCu2Te2 since these systems have been studied experimentally. The calculated Seebeck coefficients S of p-type β-BaCu2Se2 with p = 5.3 × 1019 cm−3 at 300 K are 156.7 μV K−1, and the published experimental data of β-BaCu2S2 and β-BaCu2Te2 are 216 μV K−1 and 127 μV K−1 at the same carrier concentration and temperature.9,12 At the same doping level, the Seebeck coefficient of β-BaCu2Se2 falls in between β-BaCu2S2 and β-BaCu2Te2, and this is because the electronic band structure near Fermi level gradually changes from sulfur to selenium to tellurium, which can determine the transport properties of β-BaCu2Ch2 (Ch = S, Se and Te). As shown in Fig. 5(b), electrical conductivity with respect to relaxation time σ/τ of p-type α- and β-BaCu2Se2 increase with temperature at the same doping level. | ||
| Fig. 5 Calculated electronic transport coefficients of α-BaCu2Se2 and β-BaCu2Se2 as a function of number of holes per unit cell at 300 K and 800 K. (a) Seebeck coefficient S, (b) electrical conductivity with respect to relaxation time σ/τ and (c) power factors with respect to relaxation time S2σ/τ. | ||
At the same temperature, the values σ/τ of β-BaCu2Se2 are larger than the ones of α-BaCu2Se2. This result also agrees well with the electronic band structures and effective masses of α- and β-BaCu2Se2 discussed above. The calculated power factors with respect to relaxation time S2σ/τ of p-type α-BaCu2Se2 and β-BaCu2Se2 as a function of number of holes at 300 K and 800 K are shown in Fig. 5(c). It can be seen that β-BaCu2Se2 has the higher power factor than α-BaCu2Se2, and this is mainly because β-BaCu2Se2 possesses higher electrical conductivity than α-BaCu2Se2, though it has a relatively lower Seebeck coefficients as well. In view of the natural superlattice structure of β-BaCu2Se2 that can provide intrinsically low thermal conductivity, the thermoelectric properties of tetragonal β-BaCu2Se2 is expected to perform better than orthorhombic α-BaCu2Se2, and it is worth further exploration in experiment. As can be seen from Fig. 5(c), there are peak values of S2σ/τ within the considered doping level range for the two phases, indicating that the thermoelectric performance of α- and β-BaCu2Se2 can be optimized by appropriate doping concentrations. The optimum doping concentrations of p-type α-BaCu2Se2 and β-BaCu2Se2 at 800 K based on the predicted maximum power factors are 0.425 and 0.229 holes per unit cell, respectively. The corresponding carrier concentrations are 9.16 × 1020 cm−3 and 1.99 × 1021 cm−3 for α-BaCu2Se2 and β-BaCu2Se2, respectively. In fact, when the doping exceeds 0.05 in orthorhombic α-BaCu2Se2 in experiments, the phase of tetragonal β-BaCu2Se2 will gradually appear.8 Thus, the thermoelectric performance of BaCu2Se2 can be enhanced by doping which can adjust carrier concentrations while introduce tetrahedral phase as well.
Since the Seebeck coefficients S can be directly fixed by the electronic structure without the relaxation time τ included, we will discuss the difference between theoretical and experimental S values. The Seebeck coefficients of typical β-BaCu2Se2 as a function of temperature are reported in Fig. 6. Here we only calculate Seebeck coefficients for the 30% and 35% p-type doping of β-BaCu2Se2 since these doping concentrations have been studied experimentally. As seen in Fig. 6, the Seebeck coefficient increases with increased temperature in the whole temperature range, similar to most of the thermoelectric materials.23,25–27 At the same temperature, the Seebeck coefficients of β-BaCu2Se2 decrease as the doping concentrations increases. The theoretical values are not exactly the same as the experimental results, and one possible reason for this discrepancy is the carrier concentrations of the doped β-BaCu2Se2 samples, which may slightly vary with temperature, while we set them to be constants in our calculations. The magnitude of Seebeck coefficients and their temperature dependence agree well with those observed in experiments, indicating that our calculations of transport properties are reliable.
 | ||
| Fig. 6 Calculated Seebeck coefficient S of β-BaCu2Se2 as a function of temperature. The data represented by dots are experimental measurements from ref. 8. | ||
4. Conclusions
In summary, the band structures of α-BaCu2Se2 and β-BaCu2Se2 have been investigated using first-principles calculations. It is observed that the dispersion of the upper part of the valence band near Γ point of α-BaCu2Se2 is more flat than the one of β-BaCu2Se2, and the effective masses of α-BaCu2Se2 are larger than the ones of β-BaCu2Se2, suggesting that β-BaCu2Se2 possesses the higher electrical conductivity. It is found that the upper valence band are mainly composed of Cu d and Se p orbitals from the projected DOS. The analysis of total DOS slope of the upper part of the valence band near Fermi level indicates that α-BaCu2Se2 has larger Seebeck coefficient than β-BaCu2Se2. The transport properties of p-type α- and β-BaCu2Se2 have been estimated based on semi-classical Boltzmann transport theory. Results suggest that the thermoelectric performance of tetragonal β-BaCu2Se2 is superior to orthorhombic α-BaCu2Se2. The optimal doping concentrations were estimated based on the calculated power factors as functions of the number of holes, and the Seebeck coefficient of the typical β-BaCu2Se2 were compared with experimental data with good agreement as well.Acknowledgements
The work was carried out at Shenzhen Key Laboratory of Nanobiomechanics (ZDSYS20140509162754023). We acknowledge the support by National Basic Research Program of China (973 Program, 2015CB755500), National Natural Science Foundation of China (11472236), the Natural Science Foundation of Guangdong Province (2014A030310438), China Postdoctoral Science Foundation funded project (2015M580739) and SIAT Innovation Program for Excellent Young Researchers (201405).References
- G. J. Snyder and E. S. Toberer, Nat. Mater., 2008, 7, 105 CrossRef CAS PubMed.
- X. Zhang and L. D. Zhao, Journal of Materiomics, 2015, 1, 91 Search PubMed.
- K. Biswas, J. He, I. D. Blum, C. I. Wu, T. P. Hogan, D. N. Seidman, V. P. Dravid and M. G. Kanatzidis, Nature, 2012, 489, 414 CrossRef CAS PubMed.
- B. Poudel, Q. Hao, Y. Ma, Y. Lan, A. Minnich, B. Yu, X. Yan, D. Wang, A. Muto, D. Vashaee, X. Chen, J. Liu, M. S. Dresselhaus, G. Chen and Z. Ren, Science, 2008, 320, 634 CrossRef CAS PubMed.
- J. Zhang, L. Song, G. K. Madsen, K. F. Fischer, W. Zhang, X. Shi and B. B. Iversen, Nat. Commun., 2016, 7, 10892 CrossRef CAS PubMed.
- J. Zhang, R. Liu, N. Cheng, Y. Zhang, J. Yang, C. Uher, X. Shi, L. Chen and W. Zhang, Adv. Mater., 2014, 26, 3848 CrossRef CAS PubMed.
- J. Li, L. D. Zhao, J. Sui, D. Berardan, W. Cai and N. Dragoe, Dalton Trans., 2015, 44, 2285 RSC.
- H. Zhang, S. Li, D. Li, S. Jin, S. Shen, T. Ying, Z. Lin, K. Li, D. Yuan and H. Zhao, Mater. Lett., 2015, 152, 117 CrossRef CAS.
- M. A. McGuire, A. F. May, D. J. Singh, M. H. Du and G. E. Jellison, J. Solid State Chem., 2011, 184, 2744 CrossRef CAS.
- K. Kurosaki, H. Uneda, H. Muta and S. Yamanaka, J. Alloys Compd., 2005, 388, 122 CrossRef CAS.
- K. Kurosaki, H. Uneda, H. Muta and S. Yamanaka, J. Appl. Phys., 2005, 97, 053705 CrossRef.
- K. Kurosaki, H. Uneda, H. Muta and S. Yamanaka, J. Alloys Compd., 2004, 385, 312 CrossRef CAS.
- Y. C. Wang and F. J. DiSalvo, J. Solid State Chem., 2001, 156, 44 CrossRef CAS.
- A. Assoud, Y. Cui, S. Thomas and B. Sutherland, J. Solid State Chem., 2008, 181, 2024 CrossRef CAS.
- A. Krishnapriyan, P. T. Barton, M. Miao and R. Seshadri, J. Phys.: Condens. Matter, 2014, 26, 155802 CrossRef PubMed.
- G. B. Liu, X. Q. Wang, X. J. Kuang and A. L. He, Phys. B, 2010, 405, 4582 CrossRef CAS.
- P. Jiao, C. Hu, D. Wang, Y. Zhong, H. Zhou and G. Rao, Comput. Mater. Sci., 2015, 103, 105 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- B. Hammer, L. B. Hansen and J. K. NØrskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413 CrossRef.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207 CrossRef CAS.
- E. F. Souza, C. A. Chagas, R. L. Manfro, M. M. V. M. Souza, R. B. Alencastrob and M. Schmal, RSC Adv., 2016, 6, 5057 RSC.
- L. Xi, Y. B. Zhang, X. Y. Shi, J. Yang, X. Shi, L. D. Chen, W. Zhang, J. Yang and D. J. Singh, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 155201 CrossRef.
- G. K. H. Madsen and D. J. Singh, Comput. Phys. Commun., 2006, 175, 67 CrossRef CAS.
- D. Zou, S. Xie, Y. Liu, J. Lin and J. Li, J. Mater. Chem. A, 2013, 1, 8888 RSC.
- D. Zou, Y. Liu, S. Xie, J. Lin, H. Zheng and J. Li, RSC Adv., 2014, 4, 54819 RSC.
- D. Zou, G. Nie, Y. Li, Y. Xu, H. Zheng and J. Y. Li, RSC Adv., 2015, 5, 24908 RSC.
- J. Huster and W. Bronger, Z. Anorg. Allg. Chem., 1999, 625, 2033 CrossRef CAS.
- A. Khein, D. J. Singh and C. J. Umrigar, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 4105 CrossRef CAS.
- J. Paier, R. Asahi, A. Nagoya and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 115126 CrossRef.
- D. J. Singh and I. I. Mazin, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, R1650 CrossRef CAS.
- M.-S. Lee, F. P. Poudeu and S. D. Mahanti, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 085204 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |