
Synthesis, mechanism and ethylene polymerization catalysis of Ge(iv), Sn(ii) and Zr(iv) complexes derived from substituted β-diketiminates†
Abstract
Insertion of LiCH(SiMe3)2 into the CN bond of the appropriate nitriles RCN afforded dimeric β-diketiminato lithium complexes [Li{N(SiMe3)C(R)C(H)C(R′)N(SiMe3)}]2 (1a, R = R′ = NMe2; 1b, R = But, R′ = o-C5H4N). Lithium salt 1a was used as a precursor to react with SnCl2 and GeCl4, respectively, and complexes [ClSn{N(SiMe3)C(NMe2)C(H)C(NMe2)N(SiMe3)}] (2) and [Cl2Ge{NC(NMe2)C(H)C(NMe2)N(GeCl3)}] (3) were obtained in good yields. By β-diketiminato lithium 1b ligand transfer, the cyclo-1,3-diazasilane heterocyclic complex 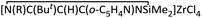 (4) was prepared using 1b and ZrCl4. Unexpectedly, the chelating β-diketiminato backbone in 3 acts as a dianionic ligand, whereas in 4, the ligand acts as a neutral cyclo-1,3-diazasilane heterocyclic ring to coordinate with the Zr center. The X-ray structures of 2–4 are presented, and reaction pathways for each complex are proposed. Upon activation with methylaluminoxane, complex 4 exhibits good activity for ethylene polymerization.
(4) was prepared using 1b and ZrCl4. Unexpectedly, the chelating β-diketiminato backbone in 3 acts as a dianionic ligand, whereas in 4, the ligand acts as a neutral cyclo-1,3-diazasilane heterocyclic ring to coordinate with the Zr center. The X-ray structures of 2–4 are presented, and reaction pathways for each complex are proposed. Upon activation with methylaluminoxane, complex 4 exhibits good activity for ethylene polymerization.

 Please wait while we load your content...
Please wait while we load your content...

