
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An easy synthetic way to exfoliate and stabilize MWCNTs in a thermoplastic pyrrole-containing matrix assisted by hydrogen bonds
R.
Araya-Hermosilla
a,
A.
Pucci
 b,
E.
Araya-Hermosilla
a,
P. P.
Pescarmona
a,
P.
Raffa
a,
L. M.
Polgar
a,
I.
Moreno-Villoslada
c,
M.
Flores
c,
G.
Fortunato
b,
A. A.
Broekhuis
a and
F.
Picchioni
*a
b,
E.
Araya-Hermosilla
a,
P. P.
Pescarmona
a,
P.
Raffa
a,
L. M.
Polgar
a,
I.
Moreno-Villoslada
c,
M.
Flores
c,
G.
Fortunato
b,
A. A.
Broekhuis
a and
F.
Picchioni
*a
aDepartment of Chemical Engineering/Product Technology, University of Groningen, Nijenborgh 4, 9747AG, Groningen, The Netherlands. E-mail: f.picchioni@rug.nl; Fax: +31-50-3634479; Tel: +31-50-363433
bDepartment of Chemistry and Industrial Chemistry, University of Pisa, Italy
cInstituto de Ciencias Químicas, Facultad de Ciencias, Universidad Austral de Chile, Chile
First published on 5th September 2016
Abstract
This work focuses on the design of an engineered thermoplastic polymer containing pyrrole units in the main chain and hydroxyl pendant groups (A-PPy-OH), which help in achieving nanocomposites containing well-distributed, exfoliated and undamaged MWCNTs. The thermal annealing at 100 °C of the pristine nanocomposite promotes the redistribution of the nanotubes in terms of a percolative network, thus converting the insulating material in a conducting soft matrix (60 μΩ m). This network remains unaltered after cooling to r.t. and successive heating cycles up to 100 °C thanks to the effective stabilization of MWCNTs provided by the functional polymer matrix. Notably, the resistivity–temperature profile is very reproducible and with a negative temperature coefficient of −0.002 K−1, which suggests the potential application of the composite as a temperature sensor. Overall, the industrial scale by which A-PPy-OH can be produced offers a straightforward alternative for the scale-up production of suitable polymers to generate multifunctional nanocomposites.
Introduction
Carbon nanotubes (CNTs) are one of the most popular fillers currently used in polymer nanocomposites (PNCs).1 CNTs are cylindrical forms of graphitic sheets displaying a single wall (SWCNT) or multi walls (MWCNT) with open or closed ends.2 CNTs are inherently multifunctional so they can work as structural supports,3 conductive4,5 and sensing platforms in PNCs.6 Conductive nanocomposites are currently used in several commercial products.7 Among them, PNCs with temperature sensing properties have become very attractive products in the open market due to their possibility of nanoscale tailoring and very low cost of production. Typically, these kinds of materials have shown a resistivity strictly dependent on temperature,8 thus opening successful applications in the field of miniaturized and potentially low cost plastic sensors.6,9–11 In some cases, the resistance variation proceeds by the dynamic interconnection/disconnection of the CNT network in the matrix.12 However, temperature sensing properties of CNTs/polymer composites are demonstrated to be more reproducible when the resistive response is governed by the semi-conducting features of exfoliated and stabilized CNT networks under thermal solicitations.11Despite all advantages of electronic temperature-sensing PNCs over conventional thermometers, these kinds of materials are not exempt of problems. A common drawback in the design of resistive sensors based on CNTs/polymer composites is the strong tendency of CNTs to aggregate in bundles during composite processing due to the strong van der Waals interactions between their graphitic surfaces, which make their large-scale utilization problematic.7,13,14
Several strategies to improve CNT dispersion in polymeric matrices have been reported in the open literature (e.g. CNT/in situ polymerization composites, high-shear melts processing, injection molding etc.15). Among them, particularly attractive is the functionalization of the CNTs surfaces by the covalent attachment of functional groups,3,15–17 but disruption of their sp2 conducting network even occurs.18,19 A non-disruptive strategy used to disperse CNTs by means of conductive polypyrroles has been reported for applications in electronics.20,21 In these particular systems, the pyrrole groups get in contact with the sp2 network of the CNTs surface via supramolecular π–π interactions, which promote the polymer wrapping around the filler. As a result, multilayers of the polymer form bridges that separate CNTs from each other yielding effective percolation pathways. However, despite the good conductive properties, polypyrroles/MWCNTs composites are highly brittle so that they must be doped with counter ions or coated on flexible polymer substrates to improve their mechanical performance (e.g. fracture toughness).22,23
In a previous work, we reported on a simple approach to cross-link MWCNTs by using polyamines synthesized from alternating aliphatic polyketone by the Paal–Knorr reaction.24 The covalent attachment of the polyamines on the MWCNTs occurred via covalent (amidation-type) grafting. The melt blending of the polyamine cross-linked MWCNTs with LDPE was attempted to reinforce the mechanical properties of the LDPE matrix. However, the enhancement of modulus and tensile strength of the LDPE matrix, as compared to un-cross-linked MWCNTs, was not achieved due to the poor dispersion of the filler.
Herein, we report on the design of a thermoplastic OH-functionalized alternating aliphatic polypyrrole (A-PPy-OH) matrix, which is capable to exfoliate and stabilize MWCNTs without the need of surface modification of the filler. The thermoplastic polymer is designed by the chemical modification of alternating aliphatic polyketone PK via Paal–Knorr reaction with amine compounds. The production of the polymer25 and its chemical modification26 occur in high yield, low cost and fast kinetic using relatively mild conditions in bulk and with water as the only by-product. Specifically, the reaction between the polymer and an OH-amine compound turns the alternating carbonyl backbone of PK into pyrrole units bearing hydroxyl moieties (Fig. 1). The combination of the polymer with MWCNTs produces a malleable and conductive rubber-like nanocomposite displaying electronic temperature-sensing properties. On one hand, the pyrrolic backbone exfoliates and stabilizes bundles of MWCNTs via supramolecular π–π interactions with the graphitic surface of the filler during thermal annealing. On the other hand, the OH-functional groups pending from the pyrrole units assist the polymer during the thermal stress by hydrogen bonding interactions in order to keep its dimensional stability and mechanical features. In order to figure out the role of the OH-motifs, a reference polymer displaying the same pyrrolic backbone of A-PPy-OH was prepared by the chemical modification of PK with n-butylamine, where the hydroxyl group is replaced by a CH3 group (A-PPy-CH3). The CNTs exfoliation and their effective dispersion within polymer matrices were investigated by in situ resistance measurements and charge-contrast SEM imaging, whereas composites resilience under thermal stress was investigated by DSC analysis. The electrical resistance of composites was eventually evaluated under thermal cycles between r.t. and 100 °C, to explore their potential for the development of sensitive, stable, and reproducible temperature sensors.
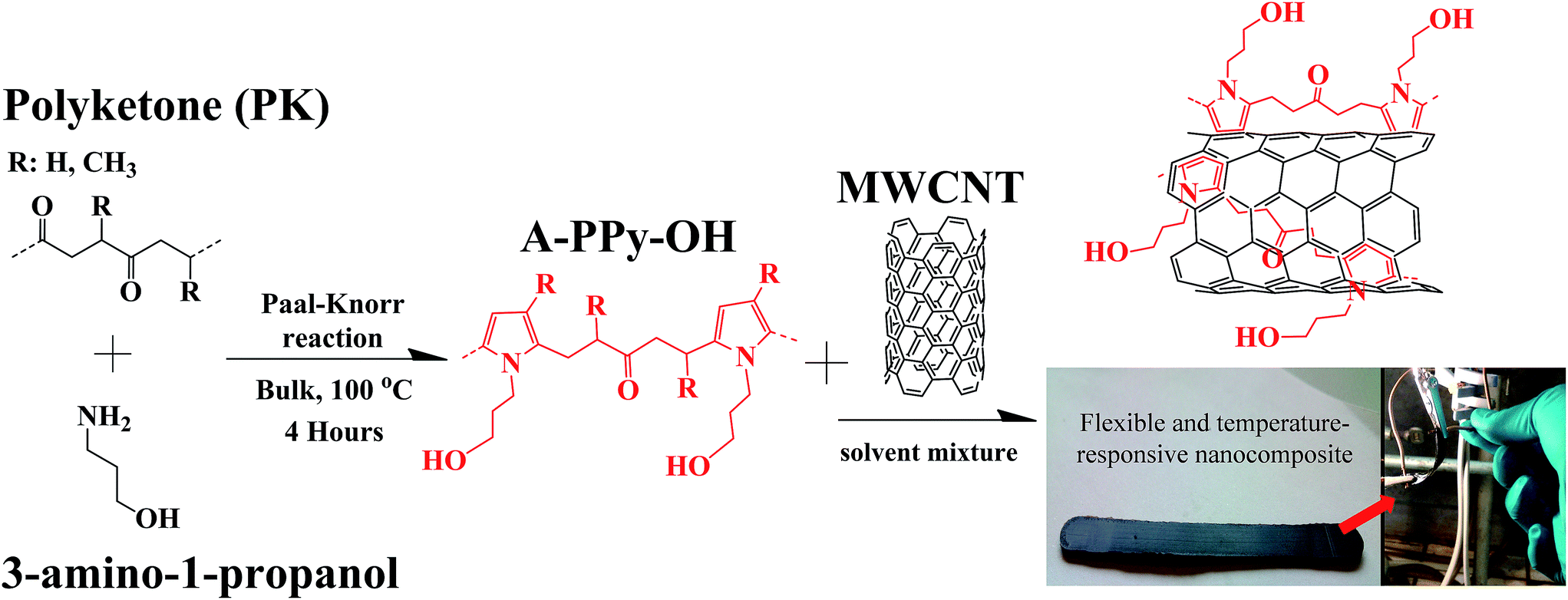 | ||
| Fig. 1 Schematic representation of PK chemically modified with 3-amino-1-propanol via the Paal–Knorr reaction (A-PPy-OH), and the resulting composite after mixing with MWCNTs. | ||
Experimental section
Materials
The alternating aliphatic polyketone (PK) was synthesized according to Mul et al. 2002.27 The resulting co- and ter-polymer of carbon monoxide presents a total olefin content of 30% of ethylene and 70% of propylene (PK30, MW 2687 Da). 3-Amino-1-propanol (OH) (Acros), butylamine (CH3) (Sigma Aldrich, 99%), 2,5-hexanedione (Sigma Aldrich 98%), multi-walled carbon nanotubes MWCNTs (O.D. 6–9 nm, L 5 μm, Sigma-Aldrich 95% carbon), DMSO-d6 (Laboratory-Scan, 99.5%), 1-methyl-2-pyrrolidinone (Sigma-Aldrich, 99.5%) were purchased and used as received.Model reaction
A model reaction between stoichiometric amounts of 2,5-hexanedione (8.7 mmol) and 3-amino-1-propanol was carried out in order to identify the presence of any side product after the Paal–Knorr reaction.26 The reaction was performed in bulk, in a 100 mL round-bottom flask equipped with a reflux condenser and a magnetic stirrer. The reaction mixture was heated up to 100 °C during 4 h.Functionalization of polyketone with alcohol pendant groups
The solvent-free Paal–Knorr reaction26 between PK and 3-amino-1-propanol was carried out using different molar ratios between the 1,4-di-carbonyl groups of polyketone and 3-amino-1-propanol (Table 1). A reference polymer that displays the same backbone structure, but bearing a CH3 instead of an OH group was also prepared using butylamine instead of 3-amino-1-propanol in the Paal–Knorr reaction, with the aim of evaluating the effect of hydrogen bonds on the thermal resilience of the composite. These chemical modifications of the polyketone were carried out in a 250 mL round-bottom glass reactor equipped with a reflux condenser, a U-type anchor impeller and an oil bath for heating. First, 60 g of PK (0.455 moles of di-carbonyl unit) were preheated to a liquid state at 100 °C. Then, 3-amino-1-propanol or butylamine was added dropwise to the reactor during 20 min. Next, the stirring speed was set to 600 rpm and the reaction was carried out for 4 h. Initially, the reaction mixture was colourless, but gradually turned to brown due to pyrrole formation on the polymer backbone.26,28,29 The resulting mixture was diluted with chloroform and washed 3 times with a 0.2 M NaCl Milli-Q water solution, to remove the unreacted 3-amino-1-propanol, if any. Thereafter, the organic phase was evaporated under vacuum at 50 °C for 24 h. Light-brown powders were obtained as final products. In order to avoid hydration, the samples were sealed in brown glass vials and stored at 6 °C for further characterization. The corresponding samples are coded as A-PPy-OHai or A-PPy-CH3ai with ai being the mol percentage of amine with respect to the carbonyl groups in the feed.| Sample | Molamine (moles) | C co a (%) | η b (%) |
|---|---|---|---|
| a C co is the % of carbonyl conversion (determined by elemental analysis). b η is the conversion efficiency of carbonyl groups. In all samples 0.455 moles of di-carbonyl unit (60 g of PK) were used. | |||
| A-PPy-OH20 | 0.091 | 15 | 75 |
| A-PPy-OH40 | 0.182 | 36 | 90 |
| A-PPy-OH60 | 0.274 | 57 | 95 |
| A-PPy-OH80 | 0.365 | 79 | 99 |
| A-PPy-CH380 | 0.365 | 78 | 98 |
The percentage of conversion of carbonyls (Cco) into pyrrole groups can be calculated as follows:
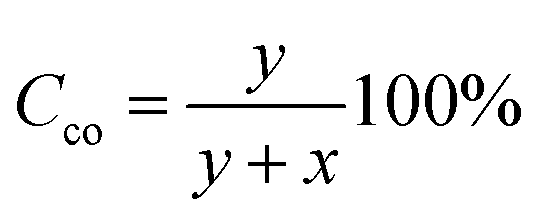
where wt(N) represents the grams of nitrogen in the final product according to elemental analysis, and Am(N) is the atomic mass of nitrogen. x can be calculated as follows:

where gprod represents the grams of product after conversion, Myw the molecular weight of the pyrrolic unit and Mpkw the molecular weight of a 1,4 di-ketone unit (131,6 g mol−1). The conversion efficiency η is defined as the ratio between the carbonyl conversion Cco and the targeted one according to the amount of polymer and amine compounds provided in the feed (Cfeedco):

the Cfeedco is calculated as follows:
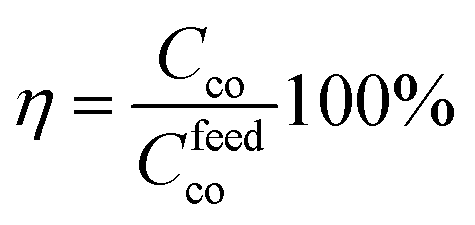
with Molamine representing the moles of amine compounds and Mold-co the moles of di-carbonyl units in the feed.

A-PPy-OH40/MWCNTs composite
A-PPy-OH40 and MWCNTs were mixed in N-methylpyrrolidone, a solvent reported as an effective dispersant for MWCNTs,16 using fixed amounts of polymer and different amounts of MWCNT expressed as wt%. For this step, only one of the OH-functionalized polymers was selected aiming at the design of a flexible temperature-responsive nanocomposite30 (Table 2).| Polymer | Polymer (g) | MWCNT (wt%) |
|---|---|---|
| A-PPy-OH40 | 4 | 2 |
| A-PPy-OH40 | 4 | 4 |
| A-PPy-OH40 | 4 | 6 |
| A-PPy-OH40 | 4 | 8 |
| A-PPy-OH40 | 4 | 10 |
In detail, 4 g of polymer were completely dissolved in N-methylpyrrolidone (10 vol%). The required wt% of MWCNTs was mixed with the same solvent and sonicated in a bath for 30 min and then poured to the polymer solution in a round bottomed flask at 50 °C for 24 h under stirring. Then, the mixture was rotary evaporated and finally transferred into a vacuum oven (80 °C for 72 h) to ensure the complete removal of the solvent. Rectangular solid samples with different dimensions were prepared by compression-molding at 150 °C for 30 minutes at 40 bar to ensure full homogeneity.
Characterization
Elemental analysis was performed with an Euro EA elemental analyzer. It was used to establish the percentage of nitrogen in the modified polymers. 1H NMR spectra were recorded on a Varian Mercury Plus 500 MHz apparatus using DMSO-d6 as solvent. ATR-FT-IR spectra were recorded using a Thermo Nicolet NEXUS 670 FT-IR. Differential scanning calorimetry DSC thermograms were recorded on a TA-Instrument DSC 2920 under N2 atmosphere. The samples were weighed (10–17 mg) in an aluminium pan, which was then sealed. The samples were first heated from −20 to 180 °C and then cooled down to −20 °C. Four cycles were performed from −20 to 180 °C, with heating and cooling rates set to 10 °C min−1. GPC measurements were performed with a HP1100 Hewlett-Packard instrument. The equipment consists of three 300 × 7.5 mm PLgel 3 μm MIXED-E columns in series and a GBC LC 1240 IR detector. The samples were dissolved in THF to obtain a final concentration of 1 mg mL−1. THF was used as eluent at a flow rate of 1 mL min−1 at 40 °C. The calibration was done using polystyrene as standard and the data were determined using PSS WinGPC software. Scanning electron microscope images were acquired on a Philips XL30 Environmental SEM FEG instrument as previously reported.31,32In situ resistance measurements were performed during thermal annealing (at 100 °C) on samples (7.28 mm long, 5.85 mm wide, 1.25 mm thick) constrained between copper plates, connected to a multimeter and placed inside a chamber provided with a heater and a temperature controller (±0.1 °C). Dynamic mechanical thermal analysis DMTA was conducted on a Rheometrics scientific solid analyzer (RSA II) under an air environment using the dual cantilever mode at an oscillation frequency of 1 Hz and a heating rate of 3 °C min−1 between 0 and 25 °C. The samples for DMTA analysis were prepared by compression-molding of 500 mg of the composite into rectangular bars (6 mm wide, 1 mm thick, 54 mm long) at 150 °C for 30 min under a pressure of 40 bar.
Results and discussion
Model component preparation
Model compounds are useful to characterize the structure of complex polymer systems via effective and rapid investigation techniques such as 1H NMR spectroscopy. Here, a model compound was prepared by reaction of 2,5-hexanedione (i.e. representative for the di-carbonyl moieties along the PK backbone) with 3-amino-1-propanol. This Paal–Knorr reaction resulted in the formation of a pyrrole unit, bearing a hydroxy terminated alkyl chain (Fig. 2). | ||
| Fig. 2 1H NMR spectrum of model compound prepared by reaction of 2,5-hexanedione with 3-amino-1-propanol. The signal of protons associated to the pyrrole ring is at 5.6 ppm, CH2 units give signals at 1.6, 3.4 and 3.8 ppm and the –OH group gives a broad signal at 4.6 ppm. | ||
Paal–Knorr chemical modification of PK
The reactions between PKs and 3-amino-1-propanol (Fig. 1) were carried out according to the different molar ratios as reported in Table 1. Notably, these functionalized PKs display a total carbonyl conversion (Cco, measured by elemental analysis) close to the target conversion (relative efficiency, η ≥ 90%, see Table 1), with a slightly lower value for the A-PPy-OH20 sample only. This result validates the robustness and versatility of the Paal–Knorr reaction of PK. The formation of the desired modified PKs containing pyrrole units with –OH motifs was confirmed by 1H NMR spectroscopy (A-PPy-OH40 sample, Fig. 3). | ||
| Fig. 3 1H NMR spectrum of A-PPy-OH40 in DMSO-d6. | ||
The 1H NMR spectrum shows analogous signals to those found in the model compound (Fig. 2), i.e. the pyrrole units at 5.8 ppm, the CH2 in the pendant group attached to the pyrrole unit at 3.5 and 3.8 ppm, and the hydroxyl group at 4.65 ppm.
An increase in the ratio between the amine compound and the polyketone in the Paal–Knorr reaction leads to the expected higher degree of conversion of the carbonyl groups of polyketone backbone into pyrrole units, as shown by the increasing Cco values in the A-PPy-OHai series from A-PPy-OH20 to A-PPy-OH80 (Table 1). The increase in the concentration of pyrrole units along the polymer backbone promotes polymer rigidity, as demonstrated by the enhancement in the Tg values of the functionalized polymers (Fig. 4).
 | ||
| Fig. 4 T g of the polymers of the A-PPy-OHai series (in black) and of A-PPy-CH380 (in red), plotted as a function of the degree of carbonyl conversion (Cco). The Tg values were measured by DSC in three temperature cycles from −20 to 180 °C. | ||
The linear increase of the Tg with Cco is not only ascribed to the presence of pyrrole units in the backbone, but mainly to effective interactions between the –OH groups of the polymer chains. This hypothesis is suggested by the much higher Tg value of A-PPy-OH80 (58 °C) compared to that of A-PPy-CH380 (14 °C), which is characterized by the same conversion degree but contains only a paraffinic moiety as pendant group. The role of –OH groups in promoting secondary interactions among macromolecules was further investigated by FT-IR spectroscopy (Fig. 5).
 | ||
| Fig. 5 FT-IR spectra of the A-PPy-OHai series and A-PPy-CH3ai. Numbers indicate final % of carbonyl conversion after Paal–Knorr reaction. | ||
The characteristic band attributed to the –OH groups stretching modes (3600–3100 cm−1) becomes progressively wider and shifts to lower energies as the degree of functionalization of the A-PPy-OHai polymers increases. This is likely due to the higher number of hydrogen donors (OH) in the polymer chains. According to the literature, hydrogen donors undergo highly directional interactions with their hydrogen acceptors.33–35 In our case and according to Fig. 5, the intensity of the peak varies with the hydrogen bonding density, which depends on the degree of polymer modification. However, if a shift of the peak is observed, it might be attributed to the balance between different competing intermolecular hydrogen bonds.33 So that, the red-shift of about 100 cm−1 observed in Fig. 5 suggests that one single directional interaction of the –OH group can be excluded. This fact can be explained by the mobility of hydrogen bonding so that its interaction with different groups on the surface of the polymer could also take place as the conversion increases and the most probable hydrogen acceptor is decreased (i.e. carbonyl groups). Moreover, we cannot exclude at this stage that the broad band at about 3400 cm−1 could be also attributed to a slight but effective hydration of the sample. In any case, the contribution of the OH groups in getting higher Tg values compared to the paraffinic counterparts results crucial since GPC measurements of all polymer systems showed no significant differences between their molecular weights (Table 3).
| Experiments | M n (×103) | M w (×103) | PDI |
|---|---|---|---|
| A-PPy-OH20 | 3.2 | 7.6 | 2.4 |
| A-PPy-OH40 | 3.0 | 7.0 | 2.3 |
| A-PPy-OH60 | 2.9 | 6.7 | 2.3 |
| A-PPy-OH80 | 2.7 | 6.2 | 2.3 |
| A-PPy-CH380 | 3.4 | 6.1 | 1.8 |
A-PPy-OH40/MWCNTs composites
Polymer nanocomposites were designed with the aim to target a flexible, light and temperature-responsive system. In this attempt, A-PPy-OH40 was chosen among the A-PPy-OHai polymers because its Tg is close to room temperature, and this is expected to provide the desirable flexibility of the composite at easily accessible temperatures. Moreover, this feature supports the use of the A-PPy-OH40/MWCNTs system as a resistive sensor for temperature variations close to the physiological regime. The A-PPy-OH40/MWCNT composites were prepared by mixing A-PPy-OH40 with different amounts of MWCNTs (Table 2) in N-methylpyrrolidone. The presence of MWCNTs causes an increase in the Tg of the composite materials compared to the parent polymer (Fig. 6). | ||
| Fig. 6 T g of the composites obtained by mixing A-PPy-OH40 with different wt% of MWCNTs. The Tg values were measured by DSC in three temperature cycles from −20 to 180 °C. | ||
According to the literature, the increase of Tg values with MWCNTs loading in polymeric systems is found to be related to the increment in their viscosity36,37 due to the interfacial interaction between matrix and filler at molecular level. Functional aromatic groups included in the backbone or as pendant groups in polymer chains, get in contact with the graphitic surface of the filler via supramolecular π–π interactions. This behavior hinders the mobility of the polymer chains and hence increases the Tg.38–40 Conductive polypyrroles have been reported as good dispersant agents for CNTs due to the interfacial connection of the filler with the pyrrole groups via π–π interactions. In our case, the backbone of the polymer contains 36% of pyrrole units that possibly promote effective interactions with the graphitic structure of CNT. In order to determine MWCNTs exfoliation, the 4 wt% MWCNT/A-PPy-OH40 nanocomposite was analyzed by SEM (Fig. 7).
 | ||
| Fig. 7 SEM morphological study of 4 wt% MWCNT/A-PPy-OH40 nanocomposite. | ||
The SEM image clearly shows the single (unbundled) nanotubes as the dominant species, thus confirming the good interactions between MWCNTs and the A-PPy-OH40 matrix.
With the aim to further improve the dispersion of the MWCNTs in the polymer matrix in terms of a fibrous percolative structure, the rubber-like composite containing 4 wt% of MWCNTs was subjected to thermal annealing at a constant temperature of 100 °C for 3 h and then cooled down to r.t. During the annealing, in situ resistivity on the compressed mold specimen was measured in order to evaluate the effect of the thermal treatment on the formation of the conductive MWCNTs network within the polymer matrix. Before annealing, the resistivity of the polymer was infinite but it decreased sharply to ≈60 μΩ m during the first minutes of thermal annealing and well persisted after cooling (Fig. 8).
 | ||
| Fig. 8 In situ electrical measurement of 4 wt% MWCNT/A-PPy-OH40 composite during thermal annealing at 100 °C. In the inset, a magnification of the first annealing instants. | ||
This behaviour indicates that thermal treatment at 100 °C effectively reorganizes MWCNTs structures within the polymer bulk in a well-defined percolative network. These remarkable changes easily occurred upon a mild thermal treatment, which is however effective in promoting polymer matrix mobility.
A morphological study of the composite analyzed by SEM in contrast mode32 corroborates the electrical measurements of the nanocomposite before and after annealing (Fig. 9). The micrographs taken from the surface of freshly teared samples revealed that bundles of MWCNTs are still present in the A-PPy-OH40 before annealing as evidenced at high magnification (Fig. 9B). It might be expected that after the sample is subjected to compression molding at 150 °C for 30 min (see experimental procedure), debundling and good dispersion of MWCNTs should be achieved. However, it is not observed in the micrograph (Fig. 9B). A possible explanation is that during compression moulding the system did not have enough time to create a well percolative network composed by debundled MWCNT. Conversely, the annealing at 100 °C, very close to the softening point, would allow the same polymer matrix mobility and provide more time to the aromatic moieties of the polymer chains to well interact with the graphitic surface thus inducing a perfect and effective exfoliation (Fig. 9C). This indicates that the thermal treatment at 100 °C favors MWCNTs debundling and promotes their homogenous dispersion in the polymer matrix (Fig. 9D) in a percolative network.
 | ||
| Fig. 9 SEM morphological study of 4 wt% MWCNT/A-PPy-OH40 composite after compression-molding at 150 °C for 30 min (A and B) and after thermal annealing (C and D) at 100 °C. | ||
The thermal history of the 4 wt% MWCNT/A-PPy-OH40 nanocomposite after thermal annealing was evaluated by DSC analysis in order to establish the resilience of the material under the investigated temperature regime (Fig. 10).
 | ||
| Fig. 10 Three consecutive DSC thermal cycles (indicated by the arrow) of 4 wt% MWCNTs/A-PPy-OH40 composite after annealing. | ||
The similarity in each consecutive thermal cycle demonstrates the resilient character of the material without any sign of thermal degradation till 180 °C. The thermal traces also indicate that the same thermodynamic response of the composite remains upon heating, thus indicating no phase separation between the components. Considering that the Tg of the composite is around 40 °C, it is worth noting that the sample does not display any endothermic transition during the annealing at 100 °C. This is another prove of the role played by the –OH groups in keeping the dimension stability of the composite.
Fig. 11 shows a picture with two different materials after being subjected to dynamic mechanical thermal analysis DMTA (from r.t. to 35 °C). A-PPy-OH40/MWCNT is capable to keep its dimensions. Contrarily, A-PPy-CH380/MWCNT gets completely deformed. This suggests that the presence of hydrogen bonding plays a relevant role in giving dimensional stability to the material.
 | ||
| Fig. 11 Pictures of two different nanocomposites displaying their differences in dimension stability (damping capability of the material) after being tested by dynamic mechanical thermal analysis DMTA (data about modulus not showed for brevity). | ||
The resistivity response of the polymer/MWCNT nanocomposite towards consecutive temperature cycles was monitored between r.t. and 100 °C (Fig. 12).
 | ||
| Fig. 12 In situ electrical measurement of 4 wt% MWCNT/A-PPy-OH40 composite during three consecutive thermal cycles between r.t. and 100 °C. | ||
Reproducible resistance variations were observed, with maximum amplitudes of 51 to 59 μohm m within the temperature interval of 80 °C. Moreover, the resistivity–temperature profile was very reproducible and with a negative temperature, thus proving that the percolation network does not experience any significant changes during the heating–cooling cycles.
The electrical response with temperature shows a negative temperature coefficient of resistance41,42 of −0.002 K−1 (average of three temperature cycles), an absolute value that is comparable to the highest values found in metals (0.0037–0.006 K−1, 0.00385 K−1 for a Pt100 sensor) and similar to other CNT/polymer nanocomposites reported in the literature.6,11,12
In summary, by this approach we have demonstrated an easy method (industrially scalable) to modulate the thermo-mechanical and electrical conductive performance of the A-PPy-OH40/MWCNT nanocomposite by adjusting the pyrrole group content and hydrogen bonding density of the polymer. Remarkable, the ability of the polymer in dispersing and stabilizing unmodified MWCNTs, via physical interactions, represents a technological advance in composite science considering the nanoscale and low cost of production at which the temperature sensor material can be tailored to be used in electronic applications.
Conclusions
We have demonstrated that a thermoplastic pyrrole-containing matrix is an effective dispersant for MWCNTs exfoliation. The polymer was prepared via the Paal–Knorr modification of an alternating aliphatic polyketone (PK) with OH-amine compound. The chemical reaction turns the waxy PK into a flexible rubber-like OH-functionalized pyrrole-containing polymer with modulable Tg depending on the amounts of –OH groups. The polymer is able to generate non-covalent functionalization of the MWCNT graphitic materials through effective π–π interactions, and that the exfoliation process does not do significant damage to the one-dimensional CNT structure. SEM micrographs and DSC traces demonstrate that the A-PPy-OH/MWCNTs nanocomposite is capable to undergo continuous thermal cycles from −20 to 180 °C without any sign of interphase modification and matrix degradation. Notably, the conductive CNT network is maintained after several temperature cycles (from r.t. to 100 °C) proving the remarkable stability of the MWCNT homogeneous dispersion within the polymer matrix. Measurements repeated over three successive heating cycles revealed highly reproducible resistivity variations with negative temperature coefficient of about −0.002 K−1, an absolute value comparable to the values found in metals. Overall, this data consistently support the use of A-PPy-OH/MWCNT nanocomposite as a soft and highly reproducible resistive sensor for temperature variations. It is certainly expected that the novelty of this approach, together with the industrial scale by which A-PPy-OH can be produced, will pave the path toward the scale-up of the proposed material at industrial level.Acknowledgements
The authors are grateful to the support of the Programa Formación de Capital Humano Avanzado, CONICYT, BECAS CHILE; grant number: 72111428. The authors are also thankful to Gert Alberda van Ekenstein, Václav Ocelik, Marcel H. de Vries, Erwin Wilbers and Anne Appeldoorn for insightful discussion and advices.Notes and references
- T. McNally and P. Pötschke, Polymer-carbon nanotube composites, Woodhead Publishing Limited, Philadelphia, USA, 2011 Search PubMed.
- S. JIMA II, Nature, 1991, 354, 56–58, DOI:10.1038/354056a0.
- M. Moniruzzaman and K. I. Winey, Macromolecules, 2006, 39, 5194–5205, DOI:10.1021/ma060733p.
- B. Wei, R. Vajtai and P. Ajayan, Appl. Phys. Lett., 2001, 79, 1172–1174, DOI:10.1063/1.1396632.
- E. Pop, D. Mann, Q. Wang, K. Goodson and H. Dai, Nano Lett., 2006, 6, 96–100, DOI:10.1021/nl052145f.
- G. Matzeu, A. Pucci, S. Savi, M. Romanelli and F. Di Francesco, Sens. Actuators, A, 2012, 178, 94–99, DOI:10.1016/j.sna.2012.02.043.
- M. F. L. De Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Science, 2013, 339, 535–539, DOI:10.1126/science.1222453.
- A. Kin-Tak Lau, in Multifunctional Polymer Nanocomposites, CRC Press, 2011, p. 19 Search PubMed.
- T. Biver, F. Criscitiello, F. Di Francesco, M. Minichino, T. Swager and A. Pucci, RSC Adv., 2015, 5, 65023–65029 RSC.
- A. Giuliani, M. Placidi, F. Di Francesco and A. Pucci, React. Funct. Polym., 2014, 76, 57–62, DOI:10.1016/j.reactfunctpolym.2014.01.008.
- N. Calisi, A. Giuliani, M. Alderighi, J. M. Schnorr, T. M. Swager, F. Di Francesco and A. Pucci, Eur. Polym. J., 2013, 49, 1471–1478, DOI:10.1016/j.eurpolymj.2013.03.029.
- H. C. Neitzert, L. Vertuccio and A. Sorrentino, IEEE Trans. Nanotechnol., 2011, 10, 688–693, DOI:10.1109/tnano.2010.2068307.
- C. Li, E. T. Thostenson and T. Chou, Compos. Sci. Technol., 2008, 68, 1227–1249, DOI:10.1016/j.compscitech.2008.01.006.
- A. Majumder, M. Khazaee, J. Opitz, E. Beyer, L. Baraban and G. Cuniberti, Phys. Chem. Chem. Phys., 2013, 15, 17158–17164, 10.1039/c3cp51844b.
- T. McNally and P. Pötschke, in Polymer–carbon nanotubes composites: preparation, properties and applications, Woodhead Publishing Limited, Cambridge, UK, 2011, p. 3 Search PubMed.
- N. Zydziak, C. Huebner, M. Bruns and C. Barner-Kowollik, Macromolecules, 2011, 44, 3374–3380, DOI:10.1021/ma200107z.
- C. Chang and Y. Liu, Carbon, 2009, 47, 3041–3049, DOI:10.1016/j.carbon.2009.06.058.
- L. Guadagno, B. De Vivo, A. Di Bartolomeo, P. Lamberti, A. Sorrentino, V. Tucci, L. Vertuccio and V. Vittoria, Carbon, 2011, 49, 1919–1930, DOI:10.1016/j.carbon.2011.01.017.
- N. G. Sahoo, S. Rana, J. W. Cho, L. Li and S. H. Chan, Prog. Polym. Sci., 2010, 35, 837–867, DOI:10.1016/j.progpolymsci.2010.03.002.
- G. Chen, M. Shaffer, D. Coleby, G. Dixon, W. Zhou, D. Fray and A. Windle, Adv. Mater., 2000, 12, 522–526 CrossRef CAS.
- Y. Su and I. Zhitomirsky, Appl. Energy, 2015, 153, 48–55, DOI:10.1016/j.apenergy.2014.12.010.
- I. M. Hodge, Method of enhancing the flexibility of polypyrrole structures, US 4642331 A, 1987.
- T. H. Lim, K. W. Oh and S. H. Kim, J. Appl. Polym. Sci., 2012, 123, 388–397, DOI:10.1002/app.34507.
- Y. Zhang, A. A. Broekhuis, M. C. A. Stuart, T. F. Landaluce, D. Fausti, P. Rudolf and F. Picchioni, Macromolecules, 2008, 41, 6141–6146 CrossRef CAS.
- E. Drent, Process for the preparation of polyketones, 1984 Search PubMed.
- Y. Zhang, A. A. Broekhuis, M. C. A. Stuart and F. Picchioni, J. Appl. Polym. Sci., 2008, 107, 262–271 CrossRef CAS.
- W. P. Mul, H. Dirkzwager, A. A. Broekhuis, H. J. Heeres, A. J. van der Linden and A. G. Orpen, Inorg. Chim. Acta, 2002, 327, 147–159 CrossRef CAS.
- C. Toncelli, D. C. De Reus, F. Picchioni and A. A. Broekhuis, Macromol. Chem. Phys., 2012, 213, 157–165 CrossRef CAS.
- Y. Zhang, A. A. Broekhuis and F. Picchioni, Macromolecules, 2009, 42, 1906–1912 CrossRef CAS.
- B. C. Tee, C. Wang, R. Allen and Z. Bao, Nat. Nanotechnol., 2012, 7, 825–832, DOI:10.1038/nnano.2012.192.
- J. Loos, A. Alexeev, N. Grossiord, C. E. Koning and O. Regev, Ultramicroscopy, 2005, 104, 160–167, DOI:10.1016/j.ultramic.2005.03.007.
- H. Deng, T. Skipa, E. Bilotti, R. Zhang, D. Lellinger, L. Mezzo, Q. Fu, I. Alig and T. Peijs, Adv. Funct. Mater., 2010, 20, 1424–1432, DOI:10.1002/adfm.200902207.
- R. M. Silverstein, G. C. Bassler and T. C. Morrill, Spectrometric identification of organic compounds, John Wiley and sons, Inc., Canada, 1991 Search PubMed.
- Y. Huang, A. K. Unni, A. N. Thadani and V. H. Rawal, Nature, 2003, 424, 146, DOI:10.1038/424146a.
- D. Seebach, A. Beck and A. Heckel, Angew. Chem., Int. Ed., 2001, 40, 92–138 CrossRef CAS.
- D. Wu, L. Wu and M. Zhang, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 2239–2251, DOI:10.1002/polb.21233.
- F. Du, R. Scogna, W. Zhou, S. Brand, J. Fischer and K. Winey, Macromolecules, 2004, 37, 9048–9055, DOI:10.1021/ma049164g.
- L. A. S. d. A. Prado, M. Kwiatkowska, S. S. Funari, Z. Roslaniec, G. Broza and K. Schulte, Polym. Eng. Sci., 2010, 50, 1571–1576, DOI:10.1002/pen.21689.
- S. Meuer, L. Braun, T. Schilling and R. Zentel, Polymer, 2009, 50, 154–160, DOI:10.1016/j.polymer.2008.10.039.
- J. Lee, S. Yoon, K. K. Kim, I. Cha, Y. J. Park, J. Choi, Y. H. Lee and U. Paik, J. Phys. Chem. C, 2008, 112, 15267–15273, DOI:10.1021/jp804485b.
- A. Naeemi and J. D. Meindl, IEEE Electron Device Lett., 2007, 28, 135–138, DOI:10.1109/LED.2006.889240.
- S. Vollebregt, S. Banerjee, K. Beenakker and R. Ishihara, IEEE Trans. Electron Devices, 2013, 60, 4085–4089, DOI:10.1109/TED.2013.2287640.
| This journal is © The Royal Society of Chemistry 2016 |