
Substrate-dependent resistance decrease of graphene by ultraviolet-ozone charge doping†
 *a
*a
Abstract
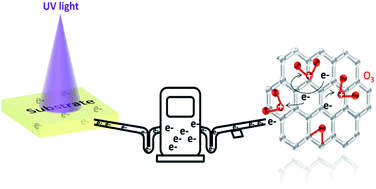
Large sheet resistance is the critical problem of graphene for application in electronic and optoelectronic devices as transparent electrodes. Ultraviolet/ozone (UVO) treatment is a convenient, highly effective, vacuum process and post-clean free method. This paper reveals that the effect of UVO treatment on the resistance of graphene is substrate dependent, which means that the band gap and photogenerated charge carriers of the substrates under UV illumination play a key role in the doping effect. The resistance of graphene can be decreased by as much as 80% on F8BT, GaN and PTFE substrates, by 70% on PMMA substrate, and by 50% on paraffin and glass substrates. Large band gap substrates (>hν) will induce a p-doping effect, while small band gap substrates (<hν) with plenty of photogenerated free charge carriers will induce n-doping effect. This approach will have great impact on the practical application of graphene in electronic and optoelectronic device fabrication.
 Please wait while we load your content...
Please wait while we load your content...

