DOI:
10.1039/C6RA07780C
(Paper)
RSC Adv., 2016,
6, 62099-62108
Mechanism of the gold(I)-catalyzed synthesis of imidazo-pyrimidines and imidazo-pyrazines via [3 + 2] dipolar cycloaddition: a DFT study†
Received
25th March 2016
, Accepted 15th June 2016
First published on 15th June 2016
Abstract
Density functional theory calculations have been carried out to study the mechanisms of gold(I)-catalyzed [3 + 2] cycloaddition reactions of pyridinium N-(heteroaryl)-aminides, as nucleophilic nitrenoids, with electron-rich alkynes, leading to imidazo-pyrimidines and imidazo-pyrazines. The calculation results suggest that the reaction mainly involves two plausible pathways leading to different products, since two carbon atoms of the asymmetric alkyne are attacked selectively by the aminide nitrogen atom. These reaction steps include C–C bond activation of the alkyne by the gold complex, a bimolecular nucleophilic attack, a closed-loop process and migration of the pyridine molecule to finally give cycloaddition products. The closed-loop process is the rate-limiting step for the whole catalytic reaction and the energy barrier shows that the pathway of the nucleophile onto the amide carbon atom of the alkyne with the electron-withdrawing group is favored. Through a detailed mechanistic investigation, we have explained the regioselectivity and the major/minor products observed by Davies et al. in gold(I)-catalyzed [3 + 2] cycloaddition reactions of nucleophilic nitrenoids with electron-rich alkynes.
1. Introduction
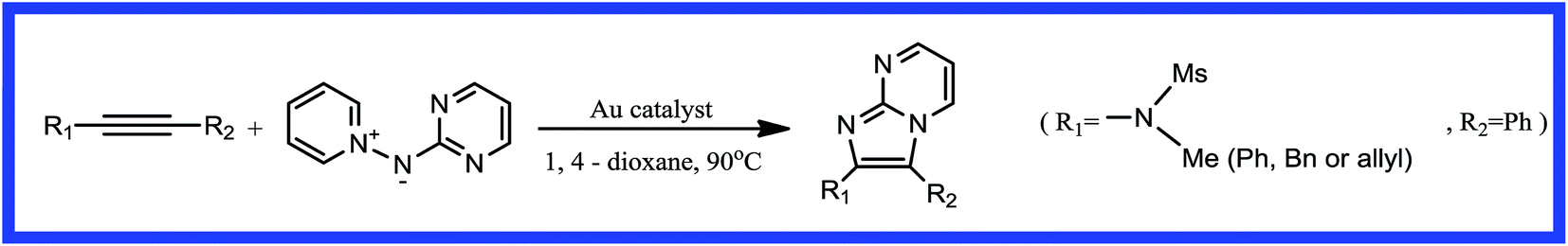
Heterocyclic compounds based on the reaction of a pyrimidine or pyrazine with the 1,2-bond of an imidazole are important components of pharmaceuticals, bioactive natural products, organic materials, and synthetic intermediates.1–5 In particular, some of their derivatives have been used as current clinical treatments and they have good pharmaceutical activity against a range of disease targets.6–9 Many methods have been developed in order to prepare these functionalized skeletons. Transition metal-catalyzed cycloadditions are among the most powerful and simple methods for the synthesis of imidazo[1,2-a] heteroaromatic frameworks from an N-nucleophilic reagent and unsaturated precursors.10–12 Gold-catalyzed dipolar cycloadditions have been widely investigated and have proven to be successful in the synthesis of various fused imidazo[1,2-a] heterocycles.13–19 In 2014, Davies et al.20 reported that the gold(I)-catalyzed [2 + 3] dipolar cycloaddition of electron-rich alkynes with pyridinium N-(heteroaryl)aminides gave imidazo-fused heteroaromatics in excellent yields in 1,4-dioxane solution at 90 °C (Scheme 1). The experimental results showed that the gold(I) complex, (ArO)3PAuCl, had the highest catalytic efficiency.
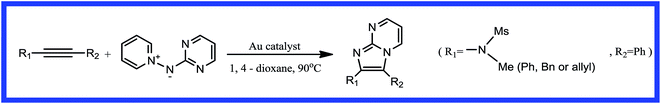 |
| | Scheme 1 Gold-catalyzed synthesis of imidazo[1,2-a] pyrimidines by [2 + 3] cycloaddition. | |
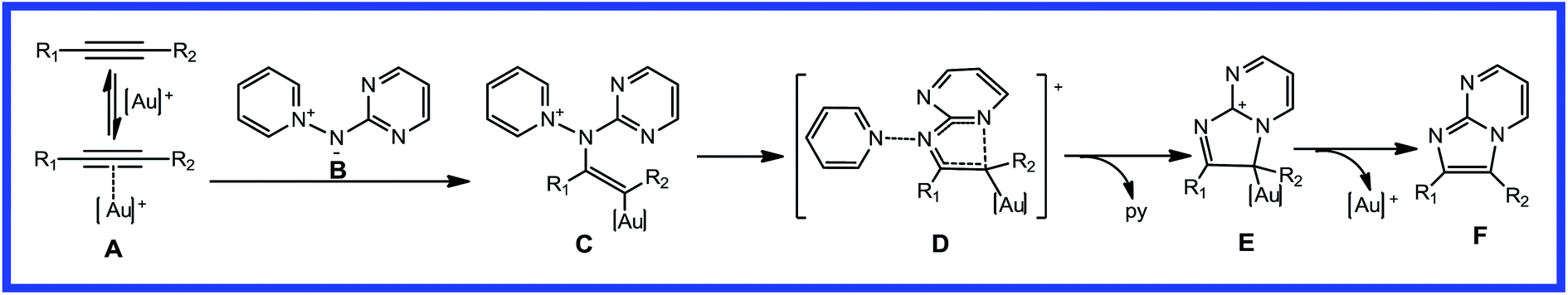
Based on experimental observations, a very brief plausible mechanism (Scheme 2) has been postulated to account for the gold(I)-catalyzed [2 + 3] cycloaddition of alkynes with aminides. In the proposed mechanism for imidazo[1,2-a]pyrimidine, cationic gold(I) first coordinates to the triple bond moiety to form the alkyne-coordinated intermediate, A. Activation of alkyne A, followed by intermolecular nucleophilic attack from B, could generate gold-carbenoid intermediate, C. Then, cyclization with elimination of pyridine gives the 5,6-ring intermediate, E. From intermediate E, the final product, F, can be formed, liberating the cationic gold(I) catalyst. The key step of this proposed mechanism is the nucleophilic attack of aminide B on the carbon atoms of the activated alkyne, A. Asymmetric alkynes may give different products, with a competitive channel and regioselectivity.
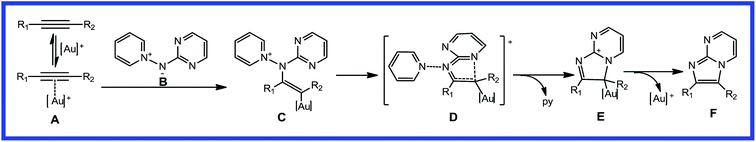 |
| | Scheme 2 Proposed catalytic cycle for gold-catalyzed [2 + 3] cycloaddition of nucleophilic nitrenoids with alkynes. | |
As far as we know, no detailed theoretical investigations for gold(I)-catalyzed [2 + 3] cycloaddition have been reported by Davies group.20 With all the interesting experimental observations given above, we carry out a detailed mechanistic investigation based on DFT computational results for the cycloaddition of pyridinium N-(heteroaryl)aminides with alkynes, using (CH3O)3PAuCl as an activating reagent. In this paper, we report the computational results and findings.
2. Computational methods
All molecular geometries reported in this paper were fully optimized in the gas phase using the M06-2X level21 of density functional theory (DFT).22 The hybrid DFT method gives reliable energies and has been successfully applied in a variety of computational studies on gold-catalyzed reactions.23–29 The effective core potential with a double-ξ valence basis set (LanL2DZ)30,31 was utilized to describe Au, and a polarization function of ξf = 1.05 was added for Au.32 The 6-31G(d,p) basis set33 was selected for all the other atoms except gold.33 Vibrational frequency calculations were done at the same level to verify the characteristics of the optimized structures as transition states or minima. All transition states in the reaction pathways were checked using the intrinsic reaction coordinate (IRC).34,35 To consider solvation-corrected and refined free energies, we employed single-point energy calculations for all of the optimized species with the double-hybrid B2PLYP/6-311++G(d,p),36 using the SMD solvation model37 in 1,4-dioxane, which was chosen as solvent for the experiments. The atomic orbital populations were calculated on the basis of NBO analyses.38 All calculations were carried out with the Gaussian 09 programs.39
3. Results and discussion
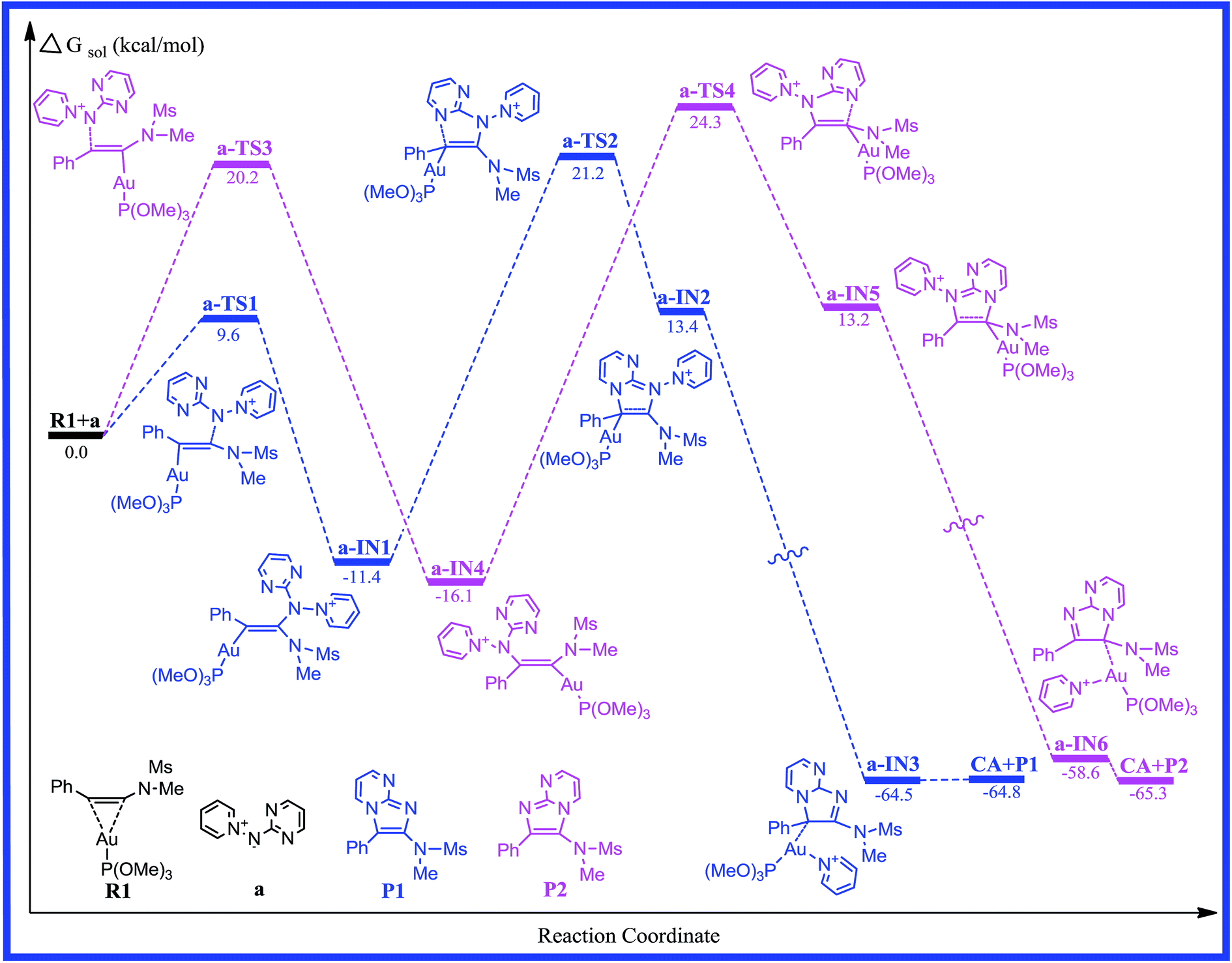
For [2 + 3] cycloaddition of alkynes with pyridinium N-(heteroaryl)aminides, catalyzed by (ArO)3PAuCl, we selected a simple model of a (CH3O)3P ligated gold(I) complex in order to maintain a low computational cost. The energy profiles for reaction pathways of gold(I)-catalyzed [2 + 3] cycloaddition are presented in Fig. 1, 3 and 5. The optimized reactants, intermediates, transition states, and products on the potential energy surfaces of the title reactions are shown in Fig. 2, 4 and 6. The detailed structural parameters for the selected stationary structures are gathered in the ESI.† We have used the Gibbs free energies obtained from the double-hybrid B2PLYP calculations in 1,4-dioxane throughout this paper unless otherwise noted.
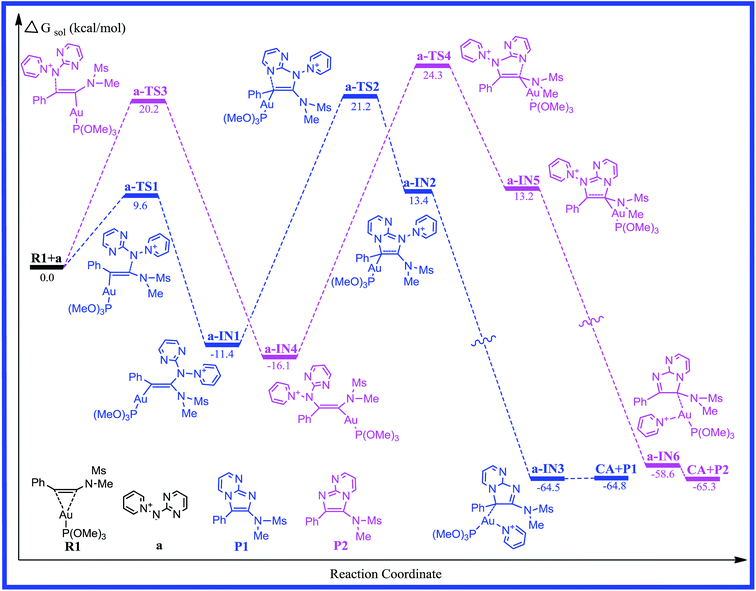 |
| | Fig. 1 Free energy profiles calculated for the gold-catalyzed [2 + 3] cycloaddition of pyridinium N-(2-pyrimidinyl) aminide with phenyl ynamide. The relative free energies are given in kcal mol−1. | |
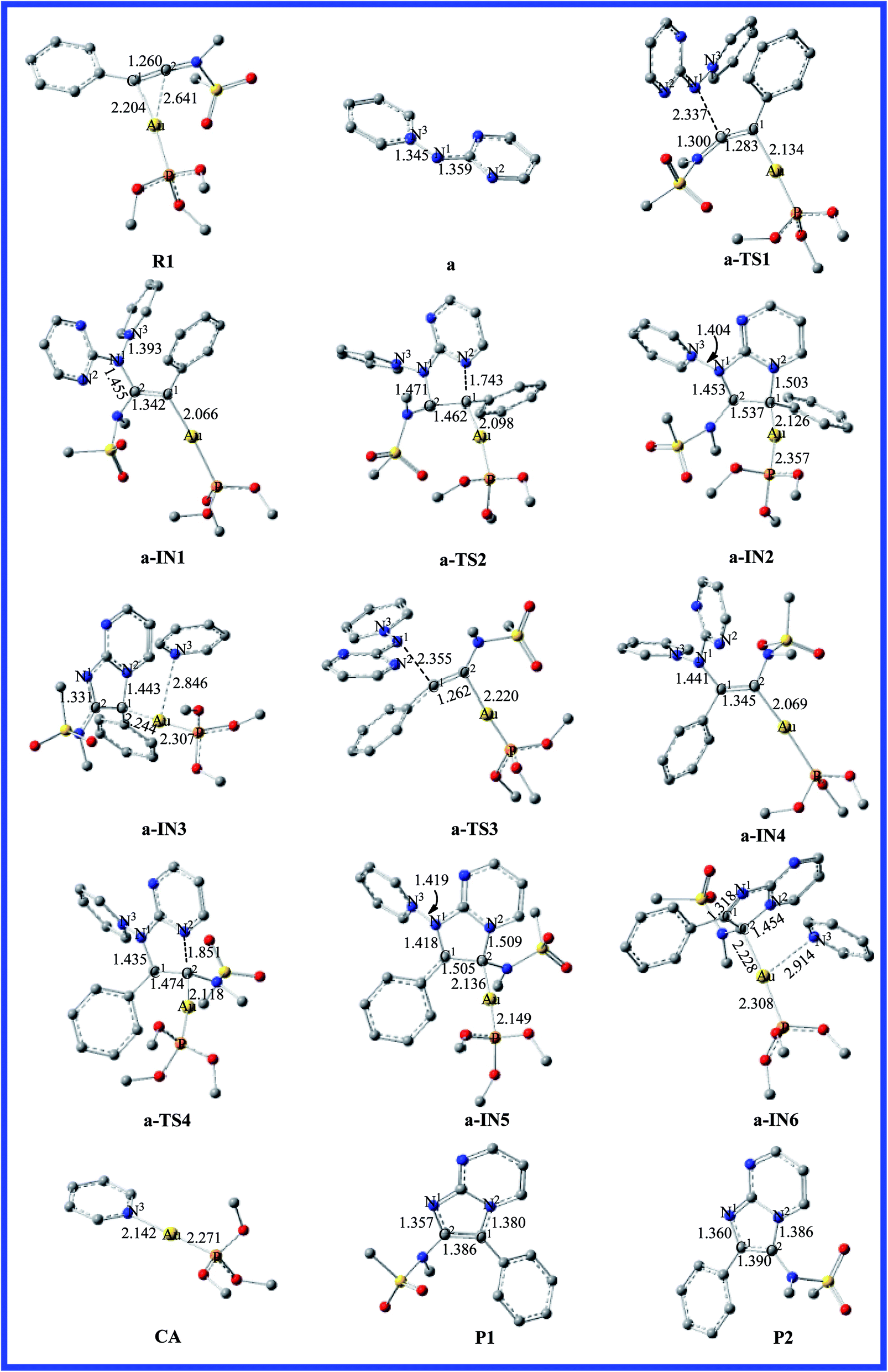
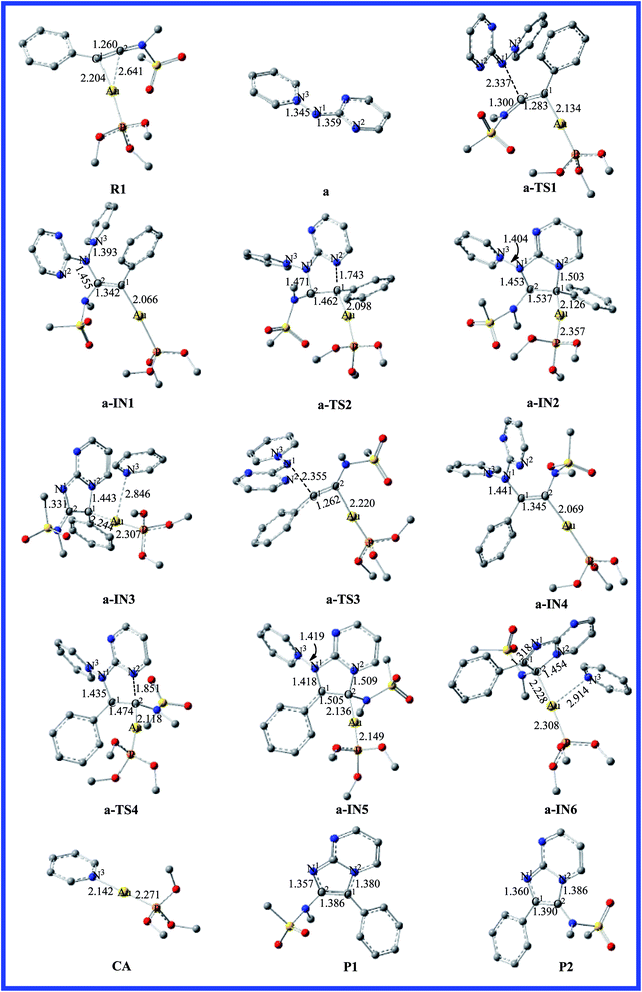 |
| | Fig. 2 Fully optimized structures of the intermediates and the transition states for pathway a are shown in Fig. 1, with selected bond lengths in Å. The H atoms in the structures are omitted for clarity. | |
3.1. Pathway a: pyridinium N-(2-pyrimidinyl) aminide with phenyl ynamide
As far as we know, the C–C triple bond functional group has two orthogonal π bonds, which have a higher energy and easily interact with the d orbitals of the electrophilic transition metal. Meanwhile, the LUMO of alkynes is easily attacked by a strong nucleophile due to its lower energy. According to the Dewar–Chatt–Duncanson (DCD) model,40–42 a study of the electronic structure and binding mode in terms of metal σ-donation and π-back-donation is important with respect to understanding the details in the literature. Once the alkyne is activated by the electrophilic metal complex, it becomes very susceptible to attack by the weaker nucleophiles.43 Thus, the interaction of the C–C triple bond of phenyl ynamide with the gold atom would give the preliminary reactant, R1. The lengths of the two Au–C bonds are 2.204 and 2.641 Å, and the C1–C2 has lost its triple-bond character and has a bond length of 1.260 Å in R1. The energy profile for the mechanism of pathway a is depicted in Fig. 1. The selected stationary structures with the key geometric parameters for the potential energy surfaces of this channel are described in Fig. 2. From pathway a, the gold-carbenoid intermediate, a-IN1, is formed through the transition structure a-TS1 when the R1 and a (pyridinium N-(2-pyrimidinyl) aminide) molecules move toward one another. Vibrational analysis shows that the a-TS1 structure has one imaginary frequency of 158.8i cm−1, and IRC calculations confirmed that this TS connects the corresponding reactants and intermediate. Fig. 2 shows that the N1 atom of molecule a attacks the C2 atom of the alkyne, the C1 atom is completely connected with the gold atom (the bond lengths N1–C2 and Au–C1 are 2.337 and 2.134 Å, respectively) in a-TS1, and the C1–C2 bond length changes from 1.260 to 1.283 Å. The vibration vector for a-TS1 is obtained and displayed by N1–C2. From changes in these structural parameters, we can see that this transition state has a similar structure to that of the intermediate, a-IN2. Examination of Fig. 1 shows that the free energy of activation for a-TS1 is calculated to be 9.6 kcal mol−1 and that for a-IN1 is −11.4 kcal mol−1 with respect to R1+a. In a-IN1, it is evident that the C1 and C2 atoms complete its sp → sp2 rehybridization, the C1–C2 bond indicates double-bond character (1.342 Å), and the N1–C2 bond formation is completed (1.455 Å). Subsequently, a-IN1 undergoes a nitrogen-transfer cyclization, resulting in the formation of intermediate a-IN2 through a-TS2 (277.4i cm−1). Fig. 2 shows that the newly formed N2–C1 bond length is 1.743 Å in a-TS2. Furthermore, the C1–C2 bond has lost its double-bond character and now has a length of 1.462 Å. The activation free energy of this step is 32.6 kcal mol−1, and the formation of a-IN2 is an endothermic process (the free energy of the formation of a-IN2 is 24.8 kcal mol−1 with respect to a-IN1), and this step is the rate-determining step of the pathway. Intermediate a-IN2 has formed a five-membered heterocyclic skeletal plane. The subsequent step of migration of the pyridine to a gold atom results in the formation of intermediate a-IN3. The formation of a-IN3 is a strongly exothermic process (the free energy of the formation of a-IN3 is −77.9 kcal mol−1 with respect to a-IN2). Finally, intermediate a-IN3, with no barrier process, generated the final product (P1), with regeneration of the catalyst (CA). From the energy profile, it is evident that the whole catalytic process is exothermic, and is −64.8 kcal mol−1 lower than the reactants (R1+a).
On the other hand, nucleophilic attack of the N2 atom of the aminide (a) at the C1 atom would lead to another possible reaction channel. The energy profile for this pathway is depicted in Fig. 1. The structures of the critical points on the potential surface, along with the essential geometric parameters, are presented in Fig. 2. In Fig. 2, the first step for the attack of N2 onto the triple bond (C1) would form new intermediate structure a-IN4 through a-TS3. The Au–C2 bond length is 2.220 Å and the N1–C1 bond length is 2.355 Å in a-TS3. Fig. 1 shows that the free energy is calculated to be 20.2 kcal mol−1 for a-TS3 and the energy for the intermediate a-IN4 is −16.1 kcal mol−1 with respect to R1+a. The higher barriers found for a-TS3 in comparison to those for a-TS1 can be explained as follows. The NBO charges for the C1, C2, and N2 atoms of the reactants (R1+a) are −0.2194, 0.3437, and −0.4339 au, respectively. The positive charge found for the C2 atom makes the nucleophilic attack of N2 on the positively charged C2 more feasible than that on the C1 atom. The formation of the N1–C1 bond in a-IN4 is completed, with a length of 1.441 Å. Furthermore, the C1–C2 bond also completed the conversion of the triple bond to a double bond, and this bond length is 1.345 Å. In order to complete the cyclization, a subsequent step gives five-membered intermediate a-IN5 through a closed-loop mechanism from a-TS4. Fig. 2 shows that the N2–C2 bond length changed from 3.222 Å in a-IN4 to 1.851 Å in a-TS4. The activation free energy for this step is 40.4 kcal mol−1, and the formation of a-IN5 is an endothermic process by 29.3 kcal mol−1. The new intermediate, a-IN6, is then obtained through pyridine transfer, which is a strongly exothermic process. Then, a-IN6 generates the final product (P2) with regeneration of the catalyst (CA). This whole reaction is exothermic by −65.3 kcal mol−1.
For the two possible channels, higher barriers are found for a-TS2 and a-TS4 and these steps are rate-determining for the whole reaction. According to the calculated results, a relatively lower barrier is found for a-TS2, while a-TS4 has a high activation barrier. The results show that the reaction system has good regioselectivity and that the major product is P1. The calculated results are consistent with the experimental results obtained by Davies et al.,20 and this reaction can be carried out only under the relevant higher reaction temperatures.
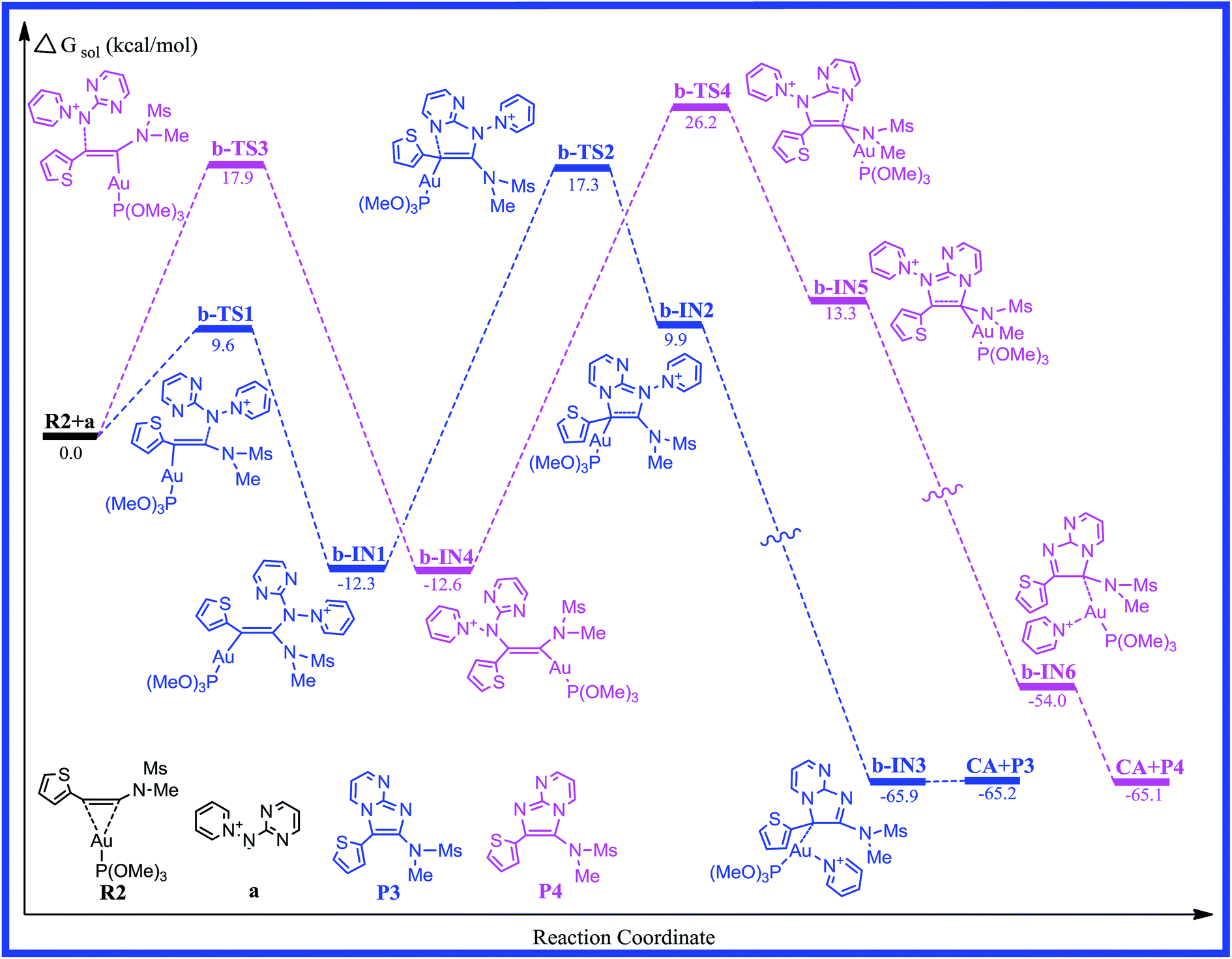
3.2. Pathway b: pyridinium N-(2-pyrimidinyl) aminide with thiophen-2-ynamide
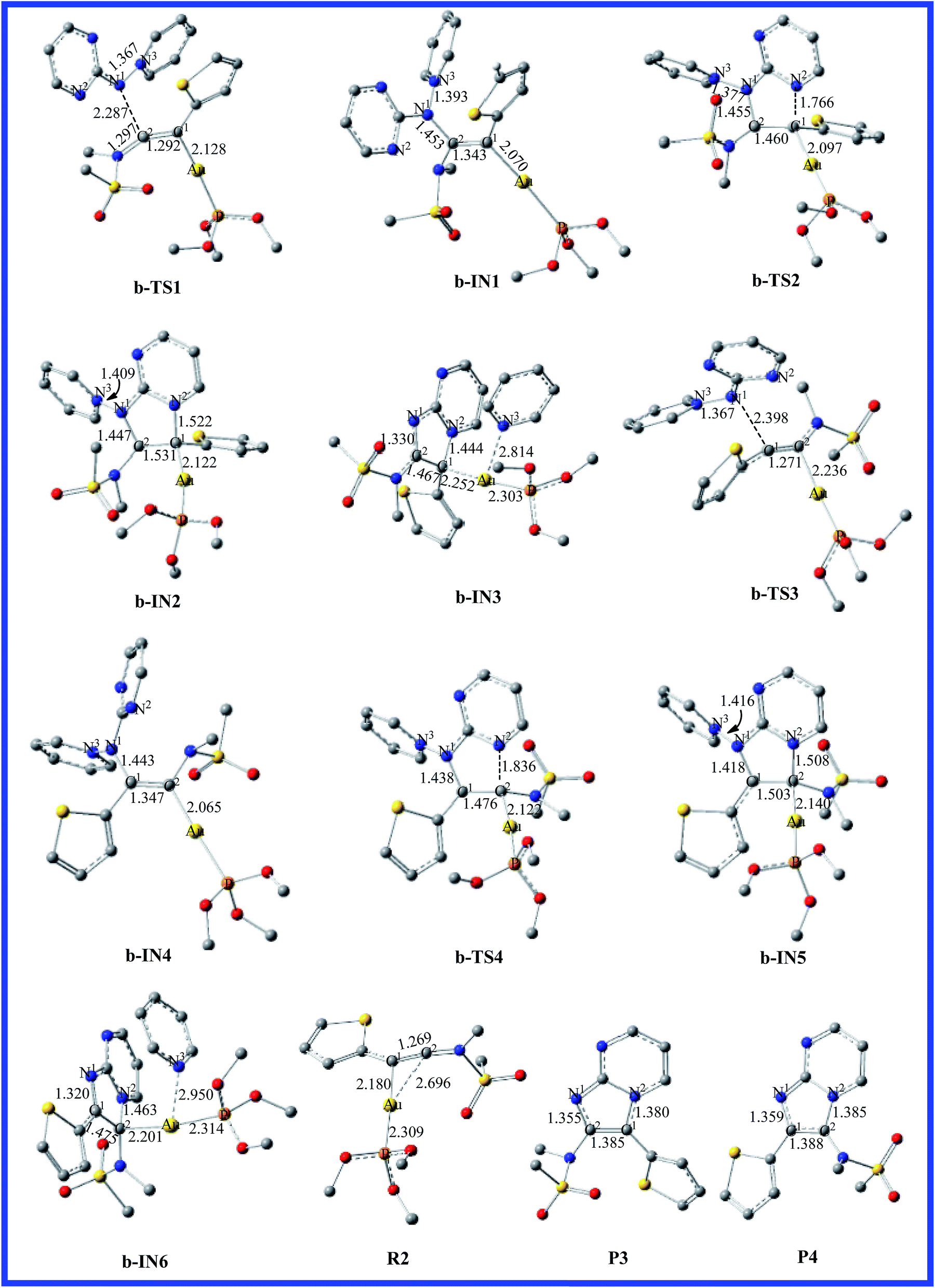
To determine more factors influencing the reaction mechanism, the authors employ aminide a, using the different ynamides to explore this transformation. The energy profile for the reaction of pyridinium N-(2-pyrimidinyl) aminide with thiophen-2-ynamide (R2) is depicted in Fig. 3. The structures, with the relevant geometric parameters on the potential surface along the reaction pathway, are presented in Fig. 4. It can be seen in Fig. 3 that the first step for pathway b involves preliminary reactant R2, stabilized by the coordination of the gold(I) atom to the π bond of the thiophen-2-ynamide. As discussed above, the coordination of the C–C triple bond with the Au atom in R2 improves the electrophilicity of the triple bond, which causes attack of the nitrogen atom onto the triple bond (C2), giving structure b-IN1 through the transition structure b-TS1, and the N1–C2 bond length is 2.287 Å in b-TS1. Fig. 3 shows that the activation free energy for this step is calculated to be 9.6 kcal mol−1 for b-TS1 and the free energy for the b-IN1 intermediate is −12.3 kcal mol−1 with respect to R2+a. The lower barrier found for b-TS1 in comparison to that for a-TS1 indicates that a thienyl group could increase the positive charge of C2 (0.3437 and 0.4911 au for R1 and R2, respectively). In principle, due to the cyclization reaction of a five-centered ring, structure b-IN1 is then converted to Au intermediate b-IN2 via the N2–C1 bond-forming transition structure, b-TS2. The free energy of activation for the second step is 29.6 kcal mol−1, and the formation of b-IN2 is endothermic by 22.2 kcal mol−1. As for pathway a, the higher energy barrier found for the second step indicates that it is also the rate-determining step for pathway b. In addition, the lower barrier found for b-TS2 in comparison to that for a-TS2 may be owing to the effect of the thienyl group. The next step for pyridine migration results in the new Au imidazole intermediate, b-IN3. Then, b-IN3, through a barrier-free process, easily generates the final product (P3), regenerating the catalyst (CA). The whole catalytic cycle is exothermic by −65.2 kcal mol−1.
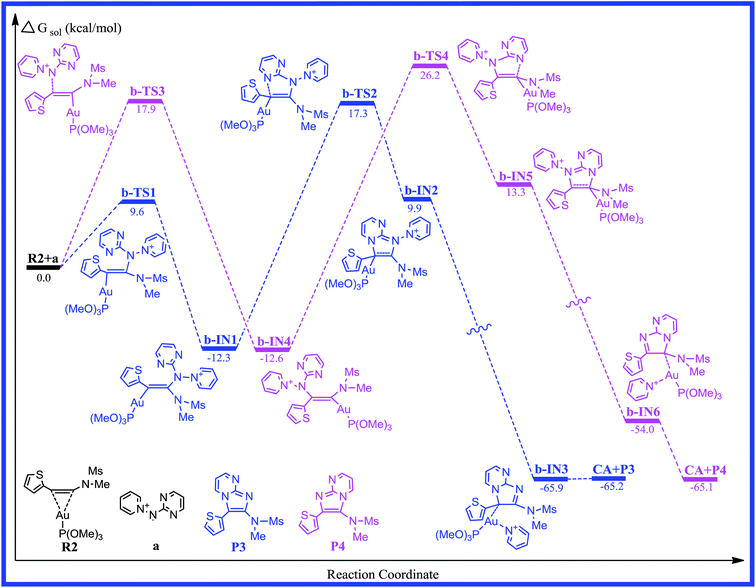 |
| | Fig. 3 Free energy profiles calculated for the gold-catalyzed [2 + 3] cycloaddition of pyridinium N-(2-pyrimidinyl) aminide with thiophen-2-ynamide. The relative free energies are given in kcal mol−1. | |
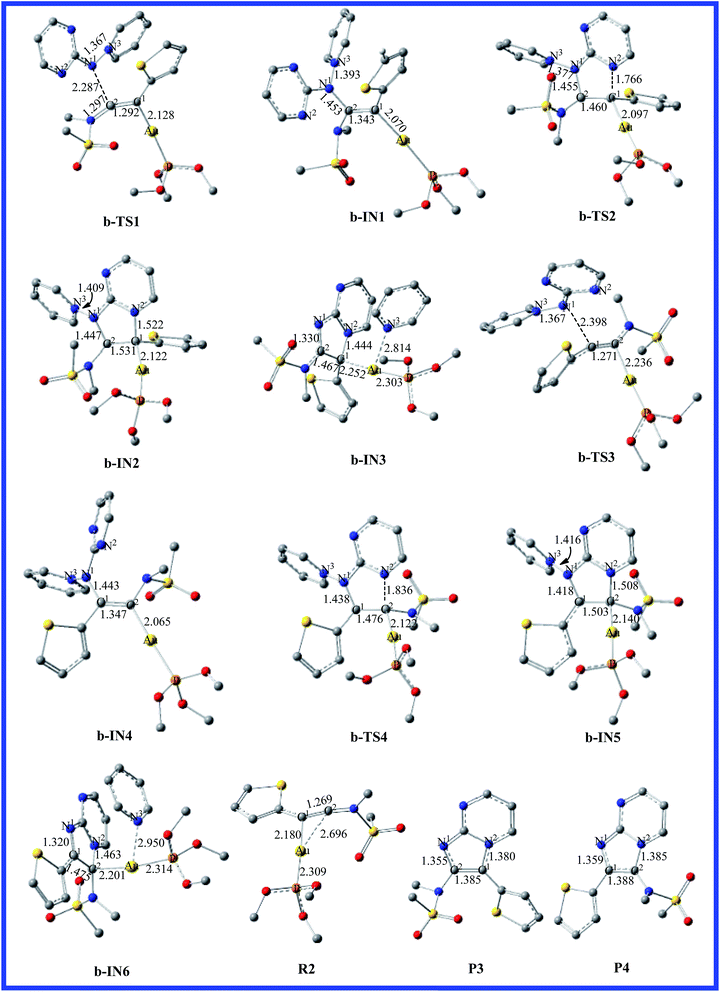 |
| | Fig. 4 Fully optimized structures of the intermediates and the transition states for pathway b are shown in Fig. 3, with selected bond lengths in Å. The H atoms in structures are omitted for clarity. | |
Reaction between the N1 atom and the C1 of the triple bond would also lead to another possible reaction channel for R2+a. In Fig. 3, as the reaction proceeds from b-TS3 to b-IN4, the N1–C1 bond formation is completed (1.443 Å). Fig. 3 shows that the free energy for b-TS3 is calculated to be 17.9 kcal mol−1 and the formation of b-IN4 is an exothermic process, which is −12.6 kcal mol−1 with respect to the reactants. Relatively lower barriers were found for b-TS3 in comparison to those for a-TS3. The C1–C2 bond has also accomplished a triple → double bond conversion and now the bond length is 1.347 Å. A subsequent step for the closed-loop process from N2 to C2 would produce the Au five-membered intermediate b-IN5 through b-TS4. Fig. 4 shows that the N2–C2 bond length changed from 1.836 in b-TS4 to 1.508 Å in b-IN5. The activation energy of this step is 38.8 kcal mol−1, and the formation of b-IN5 is an endothermic process (the energy for b-IN5 is 25.9 kcal mol−1 with respect to b-IN4). This step is also rate-determining for this reaction pathway. The last step, migration of the pyridine molecule to the Au atom forms the product (P4) and regenerates the catalyst (CA). The whole catalytic process is exothermic−65.1 kcal mol−1 lower than the preliminary reactants. The lower barrier found for b-TS2 in comparison to that for b-TS4 for the title reaction (R2+a) indicates that cyclization of the nucleophile onto the aromatic group connecting the carbon atom of the alkyne with a thienyl group is disfavored.
3.3. Pathway c: effect of the nucleophilic nitrenoids
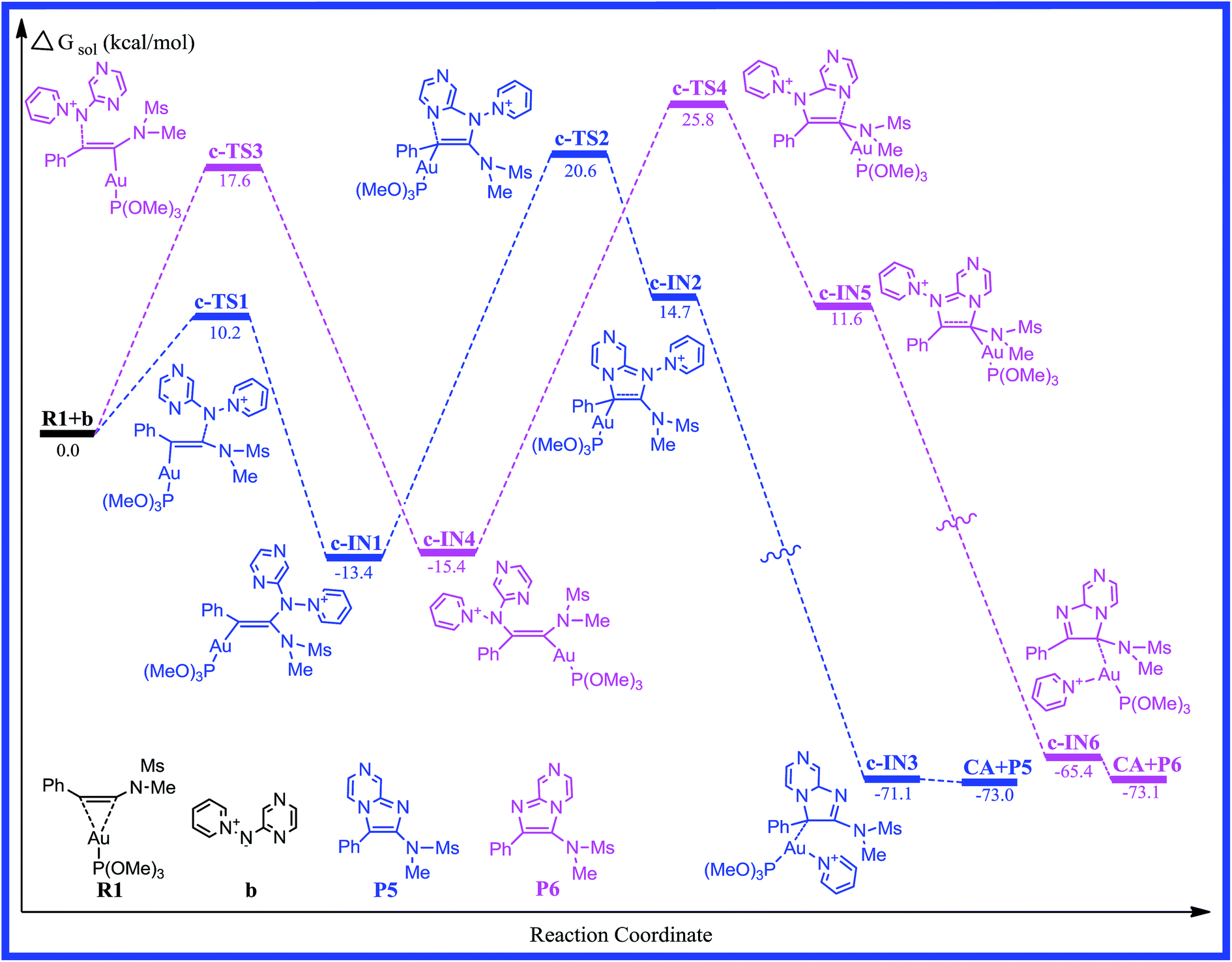
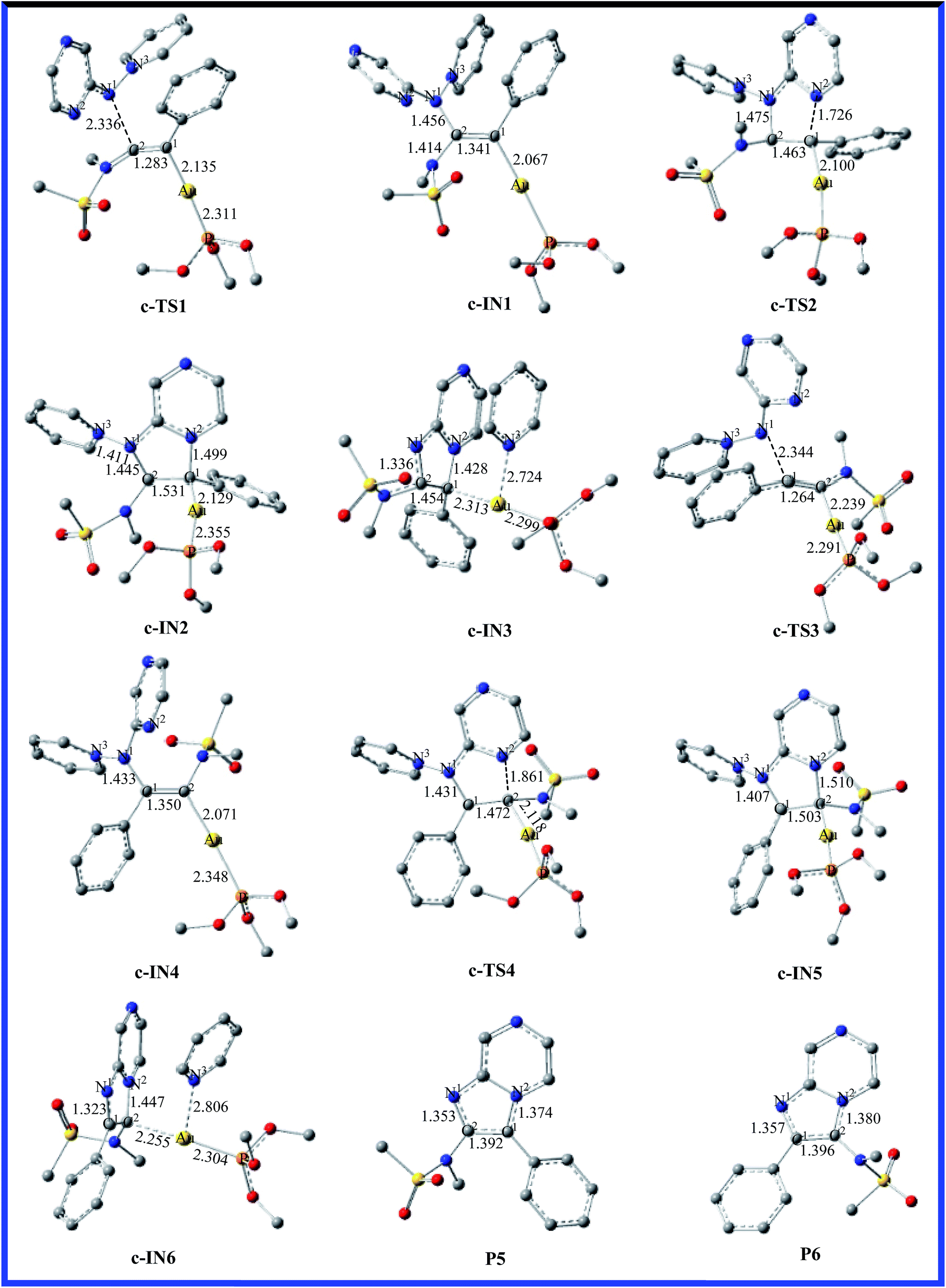
According to the computational results, the closed-loop process with the five-membered ring becomes the rate-limiting step for the title reaction system in the two possible reaction pathways. Based on the original experimental results, the authors explored the versatility of this reaction through the use of different aminides. To further explore the effect of substitution on the reaction mechanism and reactivity, reaction channel c for the pyrazinyl-substituted aminide is calculated and the energy profile for the process is shown in Fig. 5. The optimized geometries with selected key geometrical parameters (e.g., bond lengths) for the reactants (R1; b: pyridinium N-(2-pyrazinyl) aminide), intermediates, transition states, and products are depicted schematically in Fig. 6. The geometries of c-IN1-6 and c-TS1-4 are obtained, and are very similar to those of a-IN1-6, a-TS1-4 and b-IN1-6, b-TS1-4, except for the systematic difference in the structures. Fig. 5 shows that the activation free energies for c-TS1 and c-TS3 are 10.2 and 17.6 kcal mol−1. The activation free energies for c-TS2 and c-TS4 in the rate-determining step are 34.0 and 41.2 kcal mol−1, which indicates that the substitution has little influence on this pathway. A similar phenomenon is also found for pathway c. It is obvious that a lower barrier is found and the channel of R1+b → c-TS1 → c-IN1 → c-TS2 → c-IN2 → c-IN3 → CA+P5 is favorable. The calculated results are in good agreement with the experimental observations of Davies et al.20
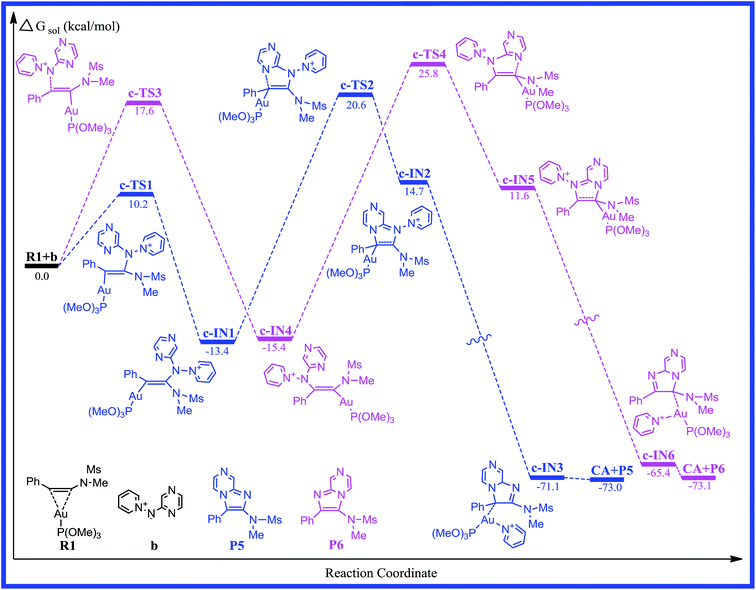 |
| | Fig. 5 Free energy profiles calculated for the gold-catalyzed [2 + 3] cycloaddition of pyridinium N-(2-pyrazinyl) aminide with phenyl ynamide. The relative free energies are given in kcal mol−1. | |
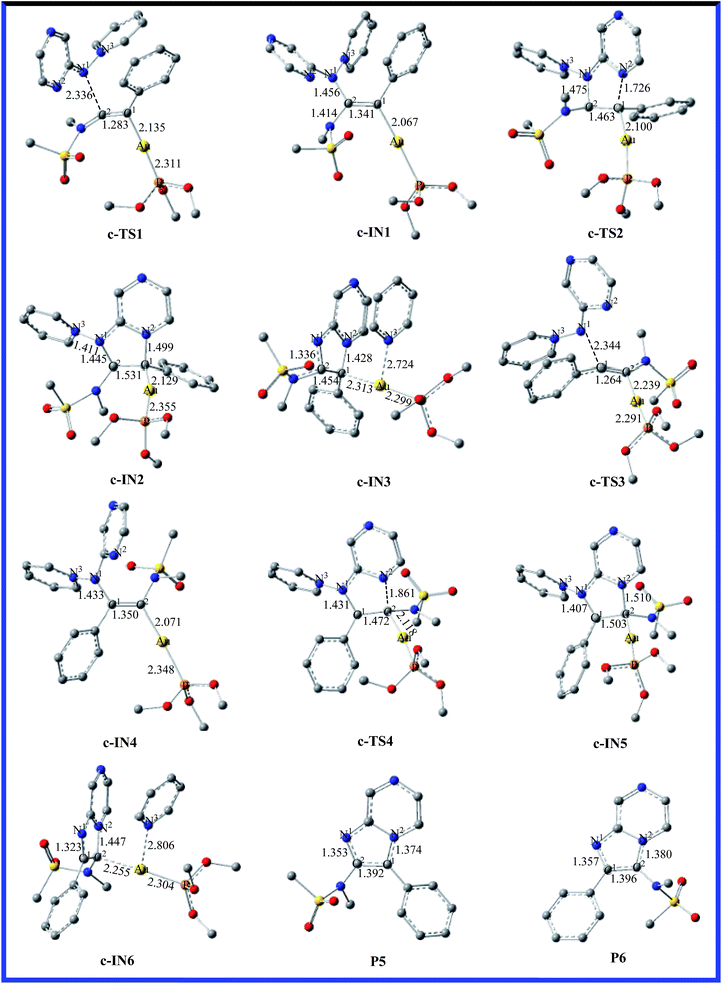 |
| | Fig. 6 Fully optimized structures of the intermediates and the transition states for pathway c are shown in Fig. 5, with selected bond lengths in Å. The H atoms in structures are omitted for clarity. | |
3.4. Attack of aromatic C or attack of amide C on nucleophilic nitrenoids
According to the experimental results, a general mechanism has been postulated to account for the formation of imidazo-pyrimidines (Scheme 2). To obtain more insight into the reaction mechanism, we carried out a detailed DFT calculation of the [3 + 2] cycloaddition reaction with the different alkynes and the different nucleophilic nitrenoids. When the alkyne is substituted with the amide group, [3 + 2] cyclization of the nucleophile onto the amide-bound carbon of the alkyne is favored, yielding intermediate C. NBO analysis shows that the positive charge found for the C2 atom makes the formation of the N1–C2 bond more feasible than that of the N1–C1 bond. The different pathways that are followed to reach a, b, c-IN1 and a, b, c-IN4 are a function of the energy difference between the transition states. According to the calculated results, the second step of the closed-loop process for a, b, c-TS2 and a, b, c-TS4 is the rate-limiting step and plays important roles in the title reaction. However, the relative energy difference between the activation free energies of TS2 and TS4 is 7.2–9.2 kcal mol−1, which suggests that the major pathway of the catalytic cycle results in the major products, P1, P3 and P5. The calculation results are also in good agreement with the experimental observations of Davies et al.
4. Conclusion
In this paper, the work has provided a theoretical investigation at the DFT (M06-2X/B2PLYP) level for the gold(I)-catalyzed [3 + 2] cycloaddition of nucleophilic nitrenoids to alkynes. The detailed aspects of the structures and energetics of the reaction pathways have been calculated. The calculations suggest that the initial catalytic cycle would include activated alkyne reactants for the gold catalyst. The first step of the reaction is the nucleophilic addition of the aminide nitrogen onto the different carbon atoms of the triple bond, to form two gold-carbenoid intermediates. Subsequently, the gold-carbenoid intermediates undergo a closed-loop process to produce the Au-imidazole intermediate, which is the rate-limiting step for the whole catalytic reaction. The observed major pathway of the nucleophile onto the amide-bound carbon of the alkyne is favored due to the electron-withdrawing group. Finally, migration of the pyridine molecule results in the formation of the final products and liberates the gold(I) catalyst. The thienyl-substituted alkyne and pyrazinyl-substituted aminide have been studied correspondingly and similar calculation results were obtained. The calculated results show that the gold-catalyzed cycloaddition is consistent with the experimental observations of Davies et al.
Acknowledgements
This study was supported by Longyuan Young Creative Talents to Support Projects, Gansu Province (2014–98), and New Chemical Materials Research and Innovation Team Project. We are grateful to the Gansu Province Supercomputer Center for essential support. We are also grateful to the reviewers for their invaluable suggestions.
Notes and references
- H. Shono, T. Ohkawa, H. Tomoda, T. Mutai and K. Araki, ACS Appl. Mater. Interfaces, 2011, 3, 654 Search PubMed.
- V. T. T. Huong, T. B. Tai and M. T. Nguyen, J. Phys. Chem. A, 2014, 118, 3335 CrossRef CAS PubMed.
- D. Davyt and G. Serra, Mar. Drugs, 2010, 8, 2755 CrossRef CAS PubMed.
- Z. Jin, Nat. Prod. Rep., 2013, 30, 869 RSC.
- W. R. Tully, C. R. Gardner, R. J. Gillespie and R. Westwood, J. Med. Chem., 1991, 34, 2060 CrossRef CAS PubMed.
- T. S. Harrison and G. M. Keating, CNS Drugs, 2005, 19, 65 CrossRef CAS.
- P. A. Bonnet, A. Michel, F. Laurent, C. Sablayrolles, E. Rechencq, J. C. Mani, M. Boucard and J. P. Chapat, J. Med. Chem., 1992, 35, 3353 CrossRef CAS PubMed.
- A. Gueiffier, M. Lhassani, A. Elhakmaoui, R. Snoeck, G. Andrei, O. Chavignon, J. C. Teulade, A. Kerbal, E. M. Essassi, J. C. Debouzy, M. Witvrouw, Y. Blache, J. Balzarini, E. De Clercq and J. P. Chapat, J. Med. Chem., 1996, 39, 2856 CrossRef CAS PubMed.
- A. N. Campbell, K. P. Cole, J. R. Martinelli, S. A. May and D. Mitchell, Org. Process Res. Dev., 2013, 17, 273 CrossRef CAS.
- E. P. A. Talbot, M. Richardson, J. M. McKenna and F. D. Toste, Adv. Synth. Catal., 2014, 356, 687 CrossRef CAS PubMed.
- Y. Gao, M. Yin, W. Wu, H. Huang and H. Jiang, Adv. Synth. Catal., 2013, 355, 2263 CrossRef CAS.
- A. Chernyak and V. Gevorgyan, Angew. Chem., Int. Ed., 2010, 49, 2743 CrossRef PubMed.
- N. Ghosh, N. Sanatan and A. K. Sahoo, Chem.–Eur. J., 2013, 19, 9428 CrossRef CAS PubMed.
- L. Li, C. Shu, B. Zhou, Y. F. Yu, X. Y. Xiao and L. W. Ye, Chem. Sci., 2014, 5, 4057 RSC.
- M. Dos Santos and P. W. Davies, Chem. Commun., 2014, 50, 6001 RSC.
- P. W. Davies, A. Cremonesi and N. Martin, Chem. Commun., 2011, 47, 379 RSC.
- Z. Xin, S. Kramer, J. Overgaard and T. Skrydstrup, Chem.–Eur. J., 2014, 20, 7926 CrossRef CAS PubMed.
- S. N. Karad and R. S. Liu, Angew. Chem., Int. Ed., 2014, 53, 9072 CrossRef CAS PubMed.
- H. V. Adcock, T. Langer and P. W. Davies, Chem.–Eur. J., 2014, 20, 7262 CrossRef CAS PubMed.
- M. Garzon and P. W. Davies, Org. Lett., 2014, 16, 4850 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- R. G. Parr and W. Yang, Density-functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
- A. Ariafard, ACS Catal., 2014, 4, 2896 Search PubMed.
- Y. Li and Z. Y. Lin, Organometallics, 2015, 34, 3538 CrossRef CAS.
- R. C. Deka, D. Bhattacharjee, A. K. Chakrabartty and B. K. Mishra, RSC Adv., 2014, 4, 5399 RSC.
- G. Ciancaleoni, L. Belpassi, D. Zuccaccia, F. Tarantelli and P. Belanzoni, ACS Catal., 2015, 5, 803 CrossRef CAS.
- X. H. Zhang and K. T. Wang, RSC Adv., 2015, 5, 34439 RSC.
- X. H. Zhang, Z. Y. Geng, Y. C. Wang, X. F. Hou and D. M. Wang, J. Mol. Catal. A: Chem., 2012, 363–364, 31 CrossRef CAS.
- R. Fang and L. Z. Yang, Organometallics, 2012, 31, 3043 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS.
- A. W. Ehlers, M. Bohme, S. Dapprich, A. Gobbi, A. Hollwarth, V. Jonas, K. F. Kohler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111 CrossRef CAS.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976 CrossRef CAS.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161 CrossRef CAS.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363 CrossRef CAS.
- G. Ciancaleoni, S. Rampino, D. Zuccaccia, F. Tarantelli, P. Belanzoni and L. Belpassi, J. Chem. Theory Comput., 2014, 10, 1021 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- E. D. Glendening, A. E. Read, J. E. Carpenter and F. Weinhold, NBO, v. 3.1, Gaussian, Inc., Pittsburgh, PA, 2009 Search PubMed.
- M. J. Frisch, et al., Gaussian 09, revision A.01, Gaussian, Inc., Pittsburgh, PA, 2009 Search PubMed.
- M. J. S. Dewar, Bull. Soc. Chim. Fr., 1951, 18, C79 Search PubMed.
- J. Chatt and L. Duncanson, J. Chem. Soc., 1953, 2939 RSC.
- D. Marchione, L. Belpassi, G. Bistoni, A. Macchioni, F. Tarantelli and D. Zuccaccia, Organometallics, 2014, 33, 4200 CrossRef CAS.
- M. García-Mota, N. Cabello, F. Maseras, A. M. Echavarren, J. Perez-Ramírez and N. Lopez, ChemPhysChem, 2008, 9, 1624 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The free energies and the activation barriers in the gas and solution phases corresponding to the relevant transition states are displayed in Table S1. Cartesian coordinates are given for optimized stationary geometries in the gas phase for all the species obtained from the DFT calculations, as well as figures giving additional structures, as noted in the text. See DOI: 10.1039/c6ra07780c |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.