
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel synthesised flavone derivatives provide significant insight into the structural features required for enhanced anti-proliferative activity†
Divyashree Ravishankara,
Kimberly A. Watsonb,
Francesca Greco*a and
Helen M. I. Osborn*a
aSchool of Pharmacy, University of Reading, Whiteknights, RG6 6AD, Reading, Berkshire, UK. E-mail: h.m.i.osborn@reading.ac.uk; f.greco@reading.ac.uk; Tel: +44 (0)118 378 7338 Tel: +44 (0)118 378 8244
bSchool of Biological Sciences, University of Reading, Harborne Building Whiteknights, RG6 6AS, Reading, Berkshire, UK
First published on 2nd June 2016
Abstract
With many cancers showing resistance to current chemotherapies, the search for novel anti-cancer agents is attracting considerable attention. Natural flavonoids have been identified as useful leads in such programmes. However, since an in-depth understanding of the structural requirements for optimum activity is generally lacking, further research is required before the full potential of flavonoids as anti-proliferative agents can be realised. Herein a broad library of 76 methoxy and hydroxy flavones, and their 4-thio analogues, was constructed and their structure–activity relationships for anti-proliferative activity against the breast cancer cell lines MCF-7 (ER +ve), MCF-7/DX (ER +ve, anthracycline resistant) and MDA-MB-231 (ER −ve) were probed. Within this library, 42 compounds were novel, and all compounds were afforded in good yields and >95% purity. The most promising lead compounds, specifically the novel hydroxy 4-thioflavones 15f and 16f, were further evaluated for their anti-proliferative activities against a broader range of cancer cell lines by the National Cancer Institute (NCI), USA and displayed significant growth inhibition profiles (e.g. compound-15f: MCF-7 (GI50 = 0.18 μM), T-47D (GI50 = 0.03 μM) and MDA-MB-468 (GI50 = 0.47 μM) and compound-16f: MCF-7 (GI50 = 1.46 μM), T-47D (GI50 = 1.27 μM) and MDA-MB-231 (GI50 = 1.81 μM)). Overall, 15f and 16f exhibited 7–46 fold greater anti-proliferative potency than the natural flavone chrysin (2d). A systematic structure–activity relationship study against the breast cancer cell lines highlighted that free hydroxyl groups and the B-ring phenyl groups were essential for enhanced anti-proliferative activities. Substitution of the 4-C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functionality with a 4-CS functionality, and incorporation of electron withdrawing groups at C-4′ of the B-ring phenyl, also enhanced activity. Molecular docking and mechanistic studies suggest that the anti-proliferative effects of flavones 15f and 16f are mediated via ER-independent cleavage of PARP and downregulation of GSK-3β for MCF-7 and MCF-7/DX cell lines. For the MDA-MB-231 cell line, restoration of the wild-type p53 DNA binding activity of mutant p53 tumour suppressor gene was indicated.
O functionality with a 4-CS functionality, and incorporation of electron withdrawing groups at C-4′ of the B-ring phenyl, also enhanced activity. Molecular docking and mechanistic studies suggest that the anti-proliferative effects of flavones 15f and 16f are mediated via ER-independent cleavage of PARP and downregulation of GSK-3β for MCF-7 and MCF-7/DX cell lines. For the MDA-MB-231 cell line, restoration of the wild-type p53 DNA binding activity of mutant p53 tumour suppressor gene was indicated.
Introduction
Natural products and their derivatives provide a significant number of new chemical leads for drug discovery programmes within a wide range of clinical areas.1 For example, analysis of plant-based compounds has highlighted many potential clinical applications for flavonoids.2 Within the area of flavonoids as anticancer agents,3–11 the success of such approaches has been exemplified through the development of flavone derivatives such as flavopiridol (for chronic lymphocytic leukemia),12–14 silibinin (for prostate cancer),15 and quercetin16 and its derivative QC12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 (for ovarian cancer), which are at various stages of clinical application. Although the natural flavonoids demonstrate anti-proliferative activities and can themselves act as useful leads, these activities have often been reported at pharmacologically non-achievable high micromolar concentrations (50–100 μM).18–21 Therefore, medicinal chemistry strategies are essential to optimise the pharmacokinetic profiles of these compounds, in order to ensure that the full clinical applications for flavonoids are realised. In this study, we have proposed methylation of hydroxyls, and conversion of 4-CO to 4-CS, as a promising strategy to improve the activities of flavonoids, since these structural motifs could lead to flavonoids with desirable physiochemical properties such as increased lipophilicity and metabolic stability. Therefore, the aim of this study was to elucidate the effects of methylation of free hydroxyls and 4-CS substitution on the anti-proliferative properties of the flavonoids by undertaking the systematic design, synthesis and biological analysis of a well-defined library of hydroxy flavone analogues. In addition, synthesis of this library of compounds would allow structure–activity relationships (SARs) to be determined. This is of particular importance for flavonoids as in-depth understanding of the SARs for anticancer properties of flavones is still limited.22,23 Specifically, the significance of some of their core structural features for activity, such as hydroxyl substitutions at different positions on the flavonoid rings, and the effect of methylation of the free hydroxyls on activity, are poorly defined. Also, the impact of modifying the 4-CO substituent on the anticancer activities of flavones has not been extensively studied.
17 (for ovarian cancer), which are at various stages of clinical application. Although the natural flavonoids demonstrate anti-proliferative activities and can themselves act as useful leads, these activities have often been reported at pharmacologically non-achievable high micromolar concentrations (50–100 μM).18–21 Therefore, medicinal chemistry strategies are essential to optimise the pharmacokinetic profiles of these compounds, in order to ensure that the full clinical applications for flavonoids are realised. In this study, we have proposed methylation of hydroxyls, and conversion of 4-CO to 4-CS, as a promising strategy to improve the activities of flavonoids, since these structural motifs could lead to flavonoids with desirable physiochemical properties such as increased lipophilicity and metabolic stability. Therefore, the aim of this study was to elucidate the effects of methylation of free hydroxyls and 4-CS substitution on the anti-proliferative properties of the flavonoids by undertaking the systematic design, synthesis and biological analysis of a well-defined library of hydroxy flavone analogues. In addition, synthesis of this library of compounds would allow structure–activity relationships (SARs) to be determined. This is of particular importance for flavonoids as in-depth understanding of the SARs for anticancer properties of flavones is still limited.22,23 Specifically, the significance of some of their core structural features for activity, such as hydroxyl substitutions at different positions on the flavonoid rings, and the effect of methylation of the free hydroxyls on activity, are poorly defined. Also, the impact of modifying the 4-CO substituent on the anticancer activities of flavones has not been extensively studied.
Results and discussion
Design and synthesis
To realise the aims of this study, structurally related hydroxy and methoxy flavones, and their 4-thio derivatives, were synthesised. The synthesis of the methoxy and hydroxy flavone scaffolds was achieved using a synthetic strategy involving the Baker–Venkataraman rearrangement as a key step (Scheme 1).24,25 The 4-thio analogues were obtained according to Scheme 226 in 20–30% overall yield (see Experimental section in ESI† material for full details).
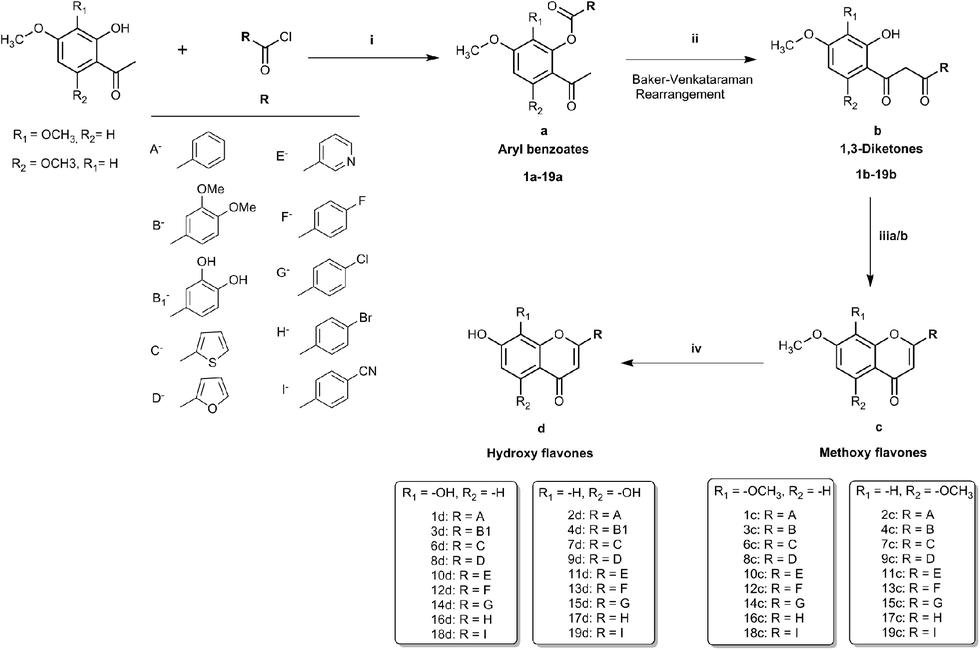 | ||
| Scheme 1 Synthesis of methoxy flavones and hydroxy flavones (1c–d to 19c–d) (i) pyridine, 75 °C, 1 h; (ii) pyridine, KOH, 50–75 °C, 2 h; (iii) (a) glacial acetic acid, 1% H2SO4, 90–110 °C, 1 h, (b) CHCl3, 0 °C to room temperature, 30 min (iv) dry DCM, BBr3, room temperature/40 °C, 4 h/overnight. | ||
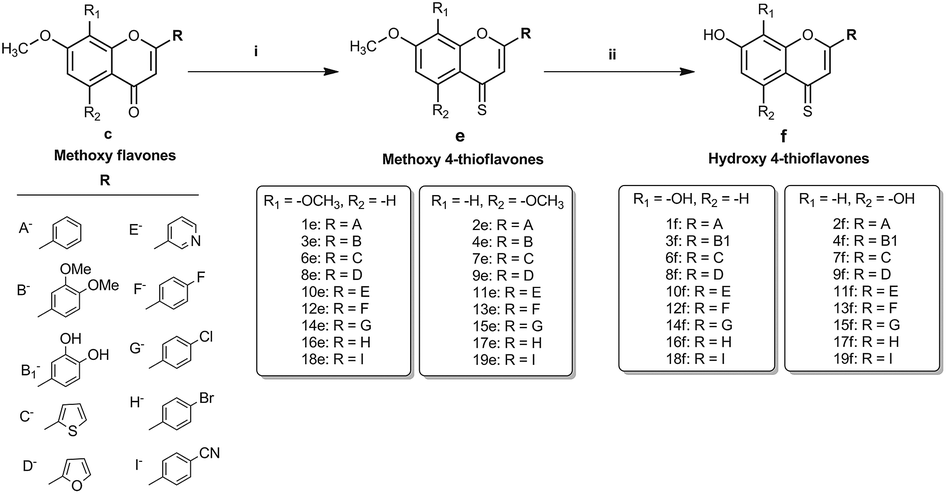 | ||
| Scheme 2 Synthesis of 4-thiocarbonyl analogues of the hydroxy and methoxy flavones (1e–f to 19e–f) (i) dry toluene, Lawesson's reagent, 110 °C, 4 h (ii) dry DCM, BBr3, room temperature/40 °C, 4 h/overnight. | ||
First, the effects of methylation and 4-thio modifications on flavones with different numbers of hydroxyl groups and different positions of hydroxyls on ring-A, were investigated. Thus, 20 flavones comprising of (i) dihydroxy flavones with hydroxyl substitution on ring-A at the C-7,8 or C-5,7 positions, (ii) tetrahydroxy flavones with dihydroxyl substitution on ring-A (at the C-7,8 or C-5,7 positions) and on ring-B (at the C-3′ and C-4′ positions), and (iii) pentahydroxy flavones with dihydroxyl substitution on ring-A (at the C-5,7 position only) and on ring-B (at the C-3′ and C-4′ positions) as well as a hydroxyl group at the C-3 position on ring-C were designed and synthesised. The analogous methoxy and 4-thio derivatives were also prepared (compounds-1c–f to 5c–f, series-1, Fig. 1).
 | ||
| Fig. 1 Library of the synthesised flavone derivatives. Novel compounds are highlighted in bold. | ||
The pentahydroxy flavone (quercetin) derivatives (5c–f) were obtained starting from pentahydroxy flavone (purchased) according to Scheme 327,28 in 36% overall yield and with 99% purity by HPLC. This represents an improvement on the recently reported synthesis of 5f, both in terms of overall yield and purity29 (22% overall yield and 95% purity). The panel of flavones studied herein also contained the well-known natural flavones chrysin (2d), luteolin (4d) and quercetin (5d) to allow comparisons to be made between the novel analogues prepared in our laboratory and some standard flavonoids. The design of further series of flavones was guided by the anti-proliferative activities of series-1 compounds. Therefore, compound-2f, a dihydroxy 4-thio flavone that was identified to possess promising anti-proliferative activity in in vitro screening (discussed below), was considered as a candidate for further optimisation through the bioisostere approach and Topliss scheme.30 This resulted in the synthesis of a focused set of dihydroxy flavone analogues (bearing C-7,8 and C-5,7 hydroxyls on ring-A) with phenyl bioisosteres (series-2, Fig. 1), and phenyl groups with electron-withdrawing substituents at the C-4′ position (based on the Topliss scheme for the optimisation of phenyl ring substitution in drug design30) (series-3, Fig. 1) as ring-B. The series-2 bioisosteric analogues of 2f comprised of dihydroxy and dimethoxy flavones, as well as their 4-thioflavone derivatives, with a five-membered 2-thienyl and 2-furanyl group, and a six-membered 3-pyridyl group [compounds 6c–f to 11c–f]. Series-3 involved 7,8 and 5,7 dihydroxy, dimethoxy flavones, and their 4-thio flavone analogues with electron-withdrawing groups (EWGs) such as fluoro (–F), chloro (–Cl), bromo (–Br) and cyano (–CN) at the C-4′ position of the phenyl ring (ring-B) [compounds 12c–f to 19c–f]. It is noteworthy that for the synthesis of novel flavones containing nitrogen atoms (e.g. the 3-pyridyl and 4-cyano phenyl groups), during the cyclisation step (step (iii-a)) in the presence of concentrated sulfuric acid in glacial acetic acid, the 1,3-diketone intermediates (compound 10b, 11b and 19b) were found to undergo hydrolysis instead of cyclisation and dehydration, to yield the corresponding methoxy flavones. Therefore, the cyclisation of such 1,3-diketones was instead accomplished in high yields (70–80%) by the addition of concentrated sulfuric acid into the solution of the 1,3-diketone in CHCl3 at 0 °C31 (step (iii-b)).
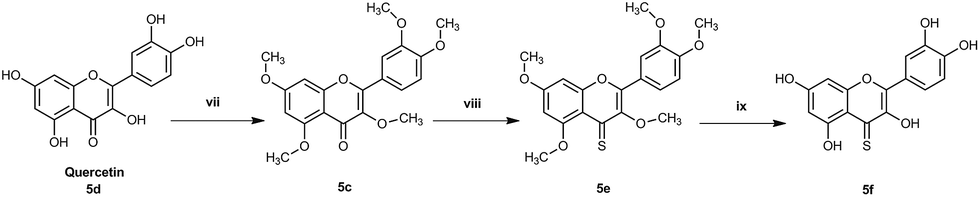 | ||
| Scheme 3 Synthesis of methoxy and 4-thiocarbonyl analogues of pentahydroxy flavone (5c–f) (i) dimethyl sulfate, 15% KOH, room temperature to 90 °C, overnight (ii) dry toluene, Lawesson's reagent, 110 °C, 4 h (iii) dry DCM, BBr3, 40 °C, overnight. | ||
Overall, multiple series of flavone derivatives comprising of hydroxy flavones (free –OH and 4-CO), hydroxy 4-thioflavones (free –OH and 4-CS), methoxy flavones (–OMe and 4-CO) and methoxy 4-thioflavones (–OMe and 4-CS) with different B-ring substitutions were successfully synthesised (summarised in Fig. 1). Out of 76 compounds, 42 compounds (3e, 3f, 4f, 6d–f, 7c–f, 8c–f, 9c, 9e, 9f, 10e, 10f, 11e, 11f, 12e, 12f, 13e, 13f, 14c–f, 15e, 15f, 16c–f, 17e, 17f, 18c, 18e, 18f, 19c, 19e and 19f) were prepared and characterised for the first time. All of the synthesised compounds were characterised by 1H and 13C NMR and IR spectroscopy, and high-resolution mass spectrometry. The purity of these compounds was established by reverse phase HPLC and the purities were found to be >95% in all cases.
Anti-proliferative evaluation
As flavonoids have been reported to show activity against breast cancer cell lines,32–34 all of the synthesised compounds (1c–f to 19c–f) were evaluated for their anti-proliferative activity against the estrogen-responsive breast cancer cell lines MCF-7, and the clinically relevant anthracycline-resistant MCF-7/DX (which mimics the multi-drug resistant scenario in patients), as well as the estrogen-independent, triple negative breast cancer cell line MDA-MB-231, using the MTT assay. The initial series of compounds (series 1, i.e. 1c–f to 5c–f) was first evaluated against MCF-7 and MCF-7/DX cell lines to investigate the effects of methylation and 4-thio substitution on flavones with differing numbers of hydroxyl groups as well as with different positions on the A-ring. The IC50 values (half maximal inhibitory concentration) of these compounds were determined as a measure of their respective cytotoxicity and these values are tabulated in Table 1. For this series, compound-2f with the dihydroxy groups at the C-5,7 positions on ring-A, and 4-thio substitution, showed the highest anti-proliferative potency with IC50 values of 7.1 ± 0.51 μM and 34.93 ± 5.75 μM against MCF-7 and MCF-7/DX cell lines, respectively. Compounds 1f, 3e, 3f, 4d and 5d showed moderate cytotoxicity with IC50 values of 25.6 ± 1.26 μM, 23.5 ± 4.27 μM and 11.8 ± 1.79 μM, respectively however, these compounds were weakly anti-proliferative against the MCF-7/DX cell line with IC50 values of 176.3 ± 1.61 μM, >250 μM and >250 μM, respectively. Compounds 1c, 1d, 1e, 9c, 3c, 3d, 4c, 4e, 5c, 5e and 5f were found to be less active against both MCF-7 and MCF-7/DX cell lines with IC50 values >30 μM and >85 μM, respectively. The IC50 values for the well-known natural flavones in this series, 2d (chrysin), 4d (luteolin) and 5d (quercetin) were found to be 25.6 ± 1.26 μM, 21.6 ± 0.81 μM and 13.7 ± 0.61 μM, respectively, against the MCF-7 cell line and the obtained values are in the range of their literature reported values.35,36 In general, all compounds exhibited a 3–10 fold lower anti-proliferative activity against MCF-7/DX, which could be because these compounds might act as substrates of the efflux pump P-gp, which is generally over expressed in the MCF-7/DX cell line.37| Chemical structure | Compound | Substitution | IC50 in MCF-7 | IC50 in MCF-7/DX |
|---|---|---|---|---|
| a Note: most active compound of the series is highlighted in bold. | ||||
 |
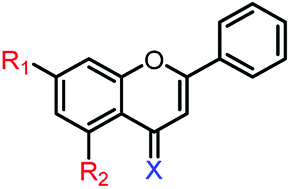
1c | R1,2 = OMe, X = O | 35.2 ± 4.4 μM | 77.0 ± 4.2 μM |
| 1d | R1,2 = OH, X = O | 38.0 ± 0.6 μM | 86.7 ± 0.9 μM | |
| 1e | R1,2 = OMe, X = S | 34.7 ± 1.7 μM | 90.3 ± 0.8 μM | |
| 1f | R1,2 = OH, X = S | 14.7 ± 1.0 μM | 47.2 ± 1.2 μM | |
 |
2c | R1,2 = OMe, X = O | 35.2 ± 4.4 μM | >250 μM |
| 2d | R1,2 = OH, X = O | 25.6 ± 1.2 μM | 176.3 ± 1.6 μM | |
| 2e | R1,2 = OMe, X = S | 23.5 ± 4.2 μM | >250 μM | |
| 2f | R1,2 = OH, X = S | 7.9 ± 0.2 μM | 34.9 ± 5.7 μM | |
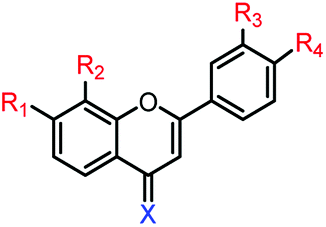 |
3c | R1,2,3,4 = OMe, X = O | >250 μM | >250 μM |
| 3d | R1,2,3,4 = OH, X = O | 97.5 ± 6.7 μM | 183.9 ± 9.0 μM | |
| 3e | R1,2,3,4 = OMe, X = S | 11.8 ± 1.7 μM | >250 μM | |
| 3f | R1,2,3,4 = OH, X = S | 16.9 ± 1.2 μM | 66.8 ± 0.7 μM | |
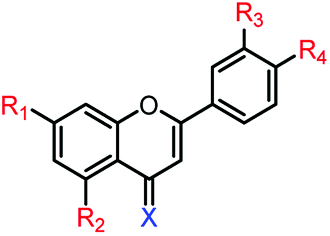 |
4c | R1,2,3,4 = OMe, X = O | >250 μM | >250 μM |
| 4d | R1,2,3,4 = OH, X = O | 21.6 ± 0.8 μM | 81.1 ± 1.7 μM | |
| 4e | R1,2,3,4 = OMe, X = S | >250 μM | >250 μM | |
| 4f | R1,2,3,4 = OH, X = S | 27.3 ± 1.5 μM | 75.9 ± 3.0 μM | |
 |
5c | R1,2,3,4,5 = OMe, X = O | 140.0 ± 1.6 μM | 245.1 ± 2.5 μM |
| 5d | R1,2,3,4,5 = OH, X = O | 13.7 ± 0.6 μM | 80.4 ± 1.7 μM | |
| 5e | R1,2,3,4,5 = OMe, X = S | >250 μM | >250 μM | |
| 5f | R1,2,3,4,5 = OH, X = S | 102.6 ± 5.3 μM | 139.3 ± 6.8 μM | |
Next, series 1 compounds (1c–f to 5c–f) were assessed for their anti-proliferative activities at 10 μM concentration against an ER −ve hormone-independent breast cancer cell line, MDA-MB-231. The 4-thio derivatives, and in particular the hydroxy compounds, exhibited greater anti-proliferative activity than the methoxy and hydroxy flavone analogues. However, no derivatives reached the IC50 at 10 μM (see ESI, Fig. S1†).
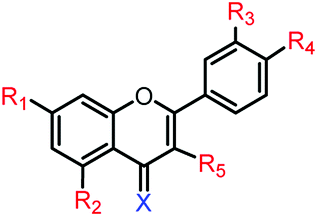
As compound-2f was identified as a promising lead compound, further novel analogues of compound-2f were prepared to discover more active flavone derivatives. Thus a series of bioisosteric analogues of the B-ring phenyl group of compound-2f (series-2, compounds 6c–f to 11c–f), and a series of compound-2f analogues with electron-withdrawing substituents at the C4′ position (series-3, compounds 12c–f to 19c–f), along with their methoxy (both 4-CO and 4-CS) and their 7,8-regioisomeric analogues (both 4-CO and 4-CS), were synthesised and evaluated for their anti-proliferative activities. Since compound-2f was identified to be active below 10 μM and 50 μM against MCF-7 and MCF-7/DX, respectively, these subsequently synthesised compounds were initially assessed for their anti-proliferative activities at 10 μM concentration against the MCF-7 and MDA-MB-231 cell lines, and at 50 μM against the MCF-7/DX cell line. Compounds that reduced the cell viability below 50% in the initial screening were arbitrarily classified as ‘active’ and they were then evaluated at a range of concentrations (between 0.1 and 100 μM) against the respective cell line to determine their IC50 values.
In the initial screening, series-2 compounds with bioisosteric replacements showed a decreased/complete loss of activity with cell viability > 50% (see ESI, Fig. S2†). Among series-3, the novel compounds 14f and 16f were found to be active against all three cell lines, and novel compounds 13f, 15f, 17f and 19f were found to be active against both the MCF-7 and MCF-7/DX cell lines. Novel compounds 12f and 18f were found to be active only against the MCF-7 cell line (Fig. 2). These active compounds were further tested against the respective cell lines at different concentrations ranging from 0.1 to 100 μM to determine their IC50 values. The determined IC50 values are listed in Table 2. Among these, novel compounds 15f (IC50 = 1.0 ± 0.1 μM and 9.1 ± 0.1 μM against MCF-7 and MCF-7/DX cell lines respectively) and 16f (IC50 = 4.9 ± 0.7 μM, 6.5 ± 0.4 μM and 8.9 ± 0.9 μM against MCF-7, MCF-7/DX and MDA-MB-231 cell lines respectively) were identified as active compounds.
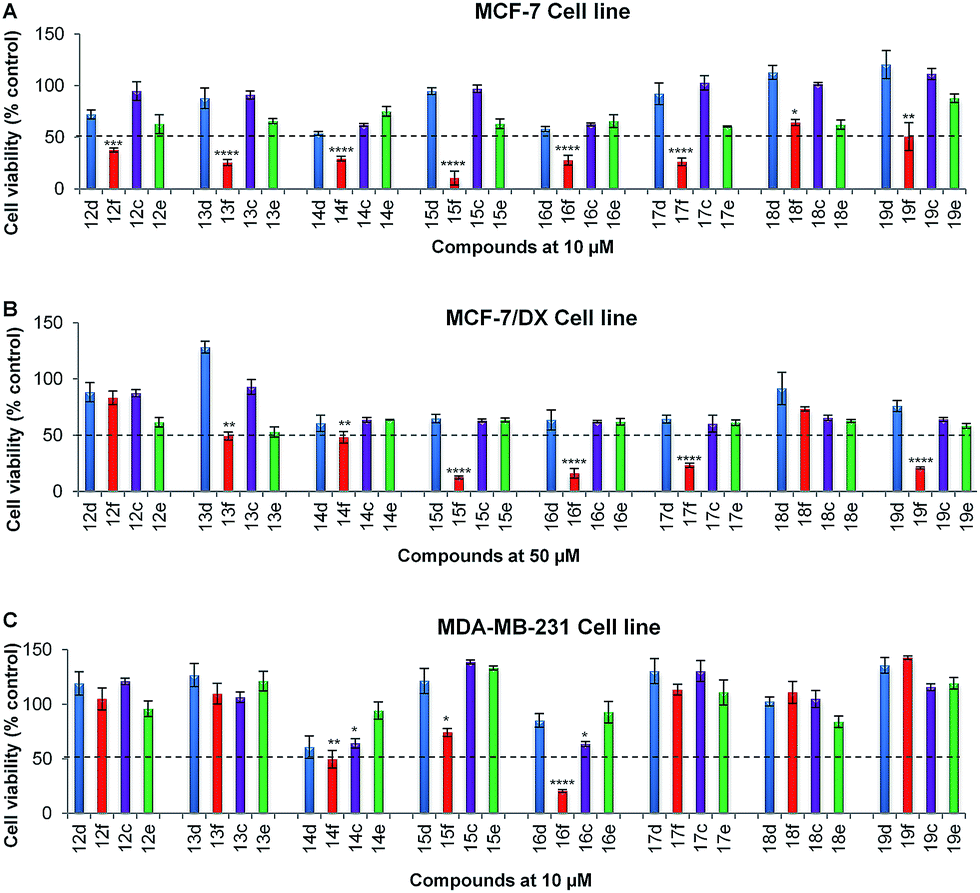 | ||
| Fig. 2 Anti-proliferative activities of compounds 12c–f to 19c–f against (A) MCF-7 cell line (B) MCF-7/DX cell line (C) MDA-MB-231 cell line. Cell viability was determined using MTT assay in the presence of compounds 12c–f to 19c–f at 10 μM concentration against the MCF-7 cell line and MDA-MB-231 cell lines, and 50 μM concentration against the MCF-7/DX cell line. Data are expressed as the mean ± standard error of the mean (SEM) (n = 3). Cells without treatment serve as control. Statistical significance was estimated, with respect to the control, by one-way ANOVA, followed by Bonferroni's post hoc test (*p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001). Dashed line corresponds to 50% cell viability and those compounds showing <50% cell viability were considered as active. Colour coding: blue – hydroxy flavone (–OH, 4-CO), red – hydroxy 4-thioflavone (–OH, 4-CS), purple – methoxy flavone (–OMe, 4-CO) and green – methoxy 4-thioflavone (–OMe, 4-CS). | ||
P valuesa
| Compound | Chemical structure | logP |
IC50 in MCF-7 | IC50 in MCF-7/DX | IC50 in MDA-MBA-231 |
|---|---|---|---|---|---|
| a Note: most active compounds of the series are highlighted in bold. Data represented is mean ± SEM, n = 3. logP values were determined using ChemDraw ultra 12.1 software. |
|||||
| 18f | 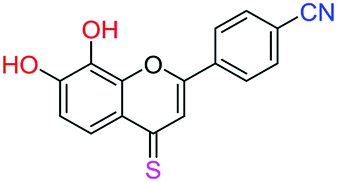 |
2.87 | 25.9 ± 1.6 μM | >50 μM | >10 μM |
| 12f | 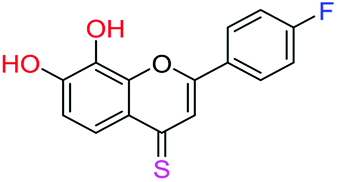 |
3.00 | 8.0 ± 0.1 μM | >50 μM | >10 μM |
| 14f | 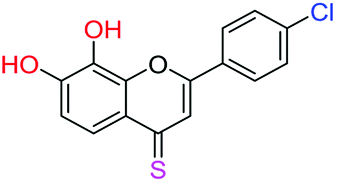 |
3.40 | 7.9 ± 2.4 μM | 34.0 ± 4.6 μM | 10.9 ± 2.4 μM |
| 16f | 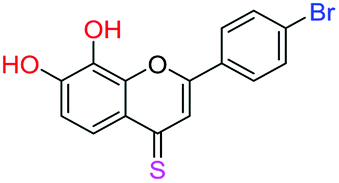 |
3.67 | 4.9 ± 0.7 μM | 6.50 ± 0.4 μM | 8.9 ± 0.8 μM |
| 19f | 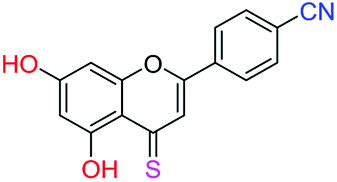 |
2.87 | 8.9 ± 0.6 μM | 27.3 ± 1.8 μM | >10 μM |
| 13f | 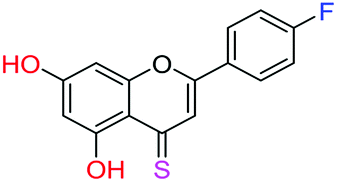 |
3.00 | 5.9 ± 0.7 μM | 41.3 ± 1.5 μM | >10 μM |
| 15f | 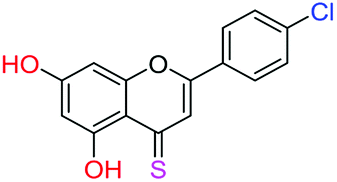 |
3.40 | 1.0 ± 0.1 μM | 9.1 ± 0.8 μM | >10 μM |
| 17f | 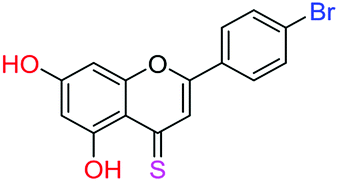 |
3.67 | 2.5 ± 0.1 μM | 15.4 ± 0.8 μM | >10 μM |
O and 4-CS) with their corresponding hydroxy flavones illustrates that the methoxy flavones were less anti-proliferative than their hydroxy analogues with the exception of compound-3e [7,8,3′,4′-tetramethoxy 4-thioflavone, IC50 = 11.8 ± 1.79 μM against the MCF-7 cell line]. For example, hydroxy flavone 4d possesses an IC50 value of 21.6 ± 0.8 μM, whereas the IC50 value of its methoxy flavone derivative-4c was found to be >250 μM. This highlights that the free hydroxyls are important for anti-proliferative activities (presumably via hydrogen bonding interactions with the potential biological cellular target).The importance of free hydroxyls for anti-proliferative activity previously has been debated in the literature, with contradicting results. Thus, the results from our programme support findings38,39 that indicate that polymethoxylation decreases the anti-proliferative activity, but contradict earlier findings that suggested higher anti-proliferative activities for polymethoxylated flavones against MCF-7,20,40 human leukemic HL-6041 and melanoma cell lines.19
Further, from the comparison of anti-proliferative activities between the hydroxy flavones (free –OH and 4-CO) and their 4-thio analogues (free –OH and 4-CS), it was apparent that the substitution of 4-CS for 4-CO enhances the anti-proliferative activity [compound-2f (free –OH and 4-CS), IC50 = 7.9 ± 0.8 μM < compound-2d (free –OH and 4-CO), IC50 = 25.6 ± 1.2 μM].
(a) Influence of number and position of hydroxyls. By comparing the anti-proliferative activities of flavone derivatives (free –OH and 4-C
O) with different numbers of hydroxy groups, it was observed that the anti-proliferative activities against the MCF-7 cell line were in the order pentahydroxy flavone (5d) [IC50 = 13.7 ± 0.6 μM] > tetrahydroxy flavone (4d) [IC50 = 21.6 ± 0.8 μM] ≈ dihydroxy flavone (2d) [IC50 = 25.6 ± 0.6 μM]. However, an opposite correlation was observed in the case of the hydroxy 4-thioflavone derivatives (free –OH and 4-CS), where the order of anti-proliferative activities was found to be dihydroxy 4-thioflavone (2f) [IC50 = 7.9 ± 0.3 μM] > tetrahydroxy 4-thioflavone (4f) [IC50 = 27.3 ± 1.5 μM] ≈ pentahydroxy 4-thioflavone (5f) [IC50 = 102.6 ± 5.4 μM]. This indicates that the number of hydroxyls is not a determining factor for the anti-proliferative activities, in the case of hydroxy flavones (free –OH and 4-CO). In complete contrast, the number of hydroxyl groups in hydroxy 4-thioflavones plays an important role in their anti-proliferative activities and the dihydroxy 4-thioflavones were found to be the most active flavones.In terms of the position of the hydroxyls, flavones with 5,7-hydroxyls on the A-ring were more active than their corresponding 7,8-hydroxy flavones. For example, 5,7,3′,4′-tetrahydroxy flavone (4d) [IC50 = 21.6 ± 0.8 μM] > 7,8,3′,4′-tetrahydroxy flavone (3d) [IC50 = 97.5 ± 6.7 μM] and 5,7-dihydroxy 4-thioflavone (2f) [IC50 = 7.9 ± 0.2 μM] > 7,8-dihydroxy 4-thioflavone (2e) [IC50 = 14.7 ± 1.0 μM].
(b) Effect of bioisosteric replacements. Replacement of the B-ring phenyl group with a bioisostere (e.g. either a five-membered 2-thienyl, a 2-furanyl group, or a six-membered 3-pyridyl group) resulted in a significant loss of anti-proliferative activity. This highlighted that the presence of a phenyl group as ring-B is favourable for anti-proliferative activity.
(c) Influence of electron-withdrawing groups. Interestingly, introduction of EWGs at the C-4′ position of the phenyl group, mainly with halogens such as F (12f and 13f), Cl (14f and 15f) and Br (16f and 17f) was found to enhance anti-proliferative activities in comparison to the flavones with an unsubstituted phenyl B-ring (compounds 1f and 2f). In particular, the introduction of Cl and Br groups increased the anti-proliferative potencies 2–7 fold (14f, 15f, 16f and 17f) against MCF-7 and MCF-7/DX cell lines, and also their introduction afforded 7,8-dihydroxy 4-thioflavones with an increased anti-proliferative activity towards MDA-MB-231 cells (compound-1f: cell viability at 10 μM was 79%, whereas, compound-14f (containing –Cl) and compound-16f (containing –Br) showed 49% and 20% cell viability at 10 μM against MDA-MB-231). The pronounced activity observed for the –F, –Cl and –Br substituted 4-thioflavones could be attributed to their increased lipophilicity, as can be observed by the decrease in IC50 values (more active) for increasing theoretical log
P values (Table 2). A summary of SARs for the anti-proliferative activities of flavones analysed herein is depicted in Fig. 3.
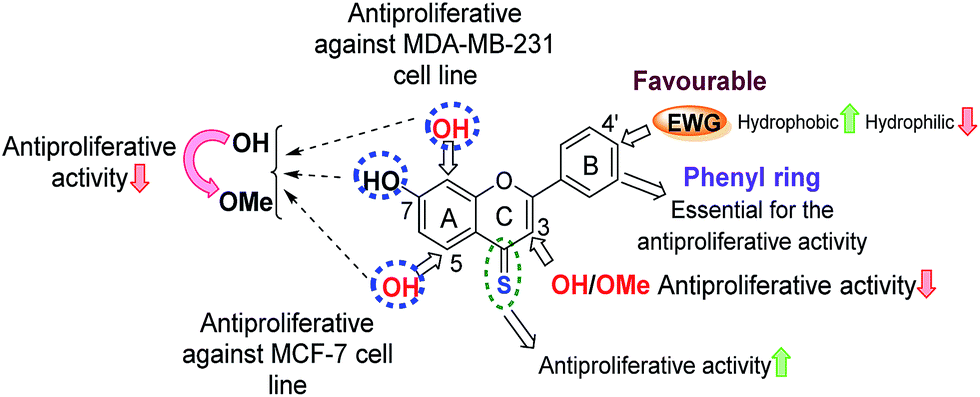 | ||
| Fig. 3 Summary of the SARs for the anti-proliferative activities of flavones analysed herein. | ||
(a) Interaction with ERα. The interaction of flavones 1c–19f with the estrogen receptor-α (ERα) was first investigated using a molecular docking approach. For this, compounds 1c–19f were docked sequentially into the binding site of the prepared X-ray crystallographic structure of human ERα complexed with an antagonist 2-phenyl-1-[4-(2-piperidin-1-yl-ethoxy)-phenyl]-1,2,3,4-tetrahydro-isoquinolin-6-ol [native ligand] (PDB code 1UOM, 2.28 Å resolution)45 using a validated docking procedure (see Experimental section). In general, no correlation was observed between the order of anti-proliferative activity and their binding affinities with ERα (as determined by their respective docking scores). One notable exception was compound-3e which showed higher binding affinity towards the ERα receptor [(docking score of 10.02, versus the native ligand docking score of 13.0), see ESI (Fig. S3†) for details]. This suggests that the majority of the flavonoid derivatives mediate their anti-proliferative effects via estrogen-independent mechanisms. A similar observation has been reported for 5-aminoflavones, which showed remarkable anti-proliferative activity against the ER +ve MCF-7 cell line without ER competition, as determined by the ER binding assay.46
To confirm this finding, an ERα antagonism assay was carried out using the human estrogen receptor alpha assay kit which consists of ERα reporter cells in which an ERα-responsive promoter is functionally linked to the luciferase reporter gene, and allows quantification of changes in the ERα activity via measurement of the changes in luciferase expression. In order to evaluate whether the flavone derivatives mediate their anti-proliferative effects via antagonism of ERα, compound-15f (IC50 = 1 μM) compound-16f (IC50 = 4.9 μM) and compound-3e (IC50 = 11.8 ± 1.79 μM), that were potent against MCF-7 cells, were assessed for their antagonist activity at four-point concentrations (10–0.1 μM) in the presence of a constant concentration of 17β-estradiol (3.2 nM, EC75). The known antagonist fulvestrant was used as a positive control. Compounds 15f and 16f did not show any decrease in the luciferase activity (induced by 17β-estradiol), which was in contrast to fulvestrant. However, compound-3e showed a dose-dependent decrease in the luciferase activity [see ESI (Fig. S4†) for details]. These results support the findings from the in silico study that the majority of these compounds (except compound-3e) most likely act through an estrogen-independent mechanism.
(b) Interactions with cell signalling proteins. Next, the signals mediated by compounds 15f and 16f in ER +ve MCF-7 and MCF-7/DX cells and ER −ve MDA-MB-231 cells were examined using the PathScan® Intracellular Signalling Array Kit, which allows simultaneous detection of 18 significant and well-characterised signalling molecules that undergo covalent post-translational modifications, such as phosphorylation and proteolysis (cleavage). Protein samples obtained from the treatment of MCF-7, MCF-7/DX and MDA-MB-231 cells with compounds 15f (IC50 < 10 μM against MCF-7 and MCF-7/DX) and 16f (IC50 < 10 μM against MCF-7, MCF-7/DX and MDA-MB-231 cells) were analysed (Fig. 4). The results showed that the anti-proliferative effects were mediated by significant inhibition of the Akt associated downstream target GSK-3β, which is a key signalling molecule for cell survival and proliferation. In addition, compound-15f, which showed greater potency towards MCF-7 cells (IC50 = 1 μM for 15f vs. IC50 = 4.9 μM for 16f), was found to trigger cell death through PARP cleavage, which is a hallmark of the cell death pathway.47 This explains the greater potency of compound-15f towards MCF-7 cells. It is noteworthy that no caspase-3 cleavage was observed; this suggests that the observed PARP cleavage is caspase-3 independent and the activation of PARP cleavage in MCF-7 cells might have been executed via other caspases such as caspases-6/9.48 In the case of MCF-7/DX cells, compound-16f, that has greater activity towards MCF-7/DX cells than compound-15f, mediated its anti-proliferative effects through a significant reduction in the phosphorylation of several cell survival proteins such as mTOR, GSK-3β and Stat3, along with induction of cell death through caspase-3 and PARP cleavage. However, compound-15f was found to elicit its anti-proliferative effects through a reduction in the phosphorylation of GSK-3β as well as through induction of cell death via PARP cleavage. Thus, the higher potency of compound-16f could be related to its ability to downregulate multiple cell survival proteins (mTOR, GSK-3β and Stat3) along with its ability to induce cell death via caspase-3 and PARP cleavage.
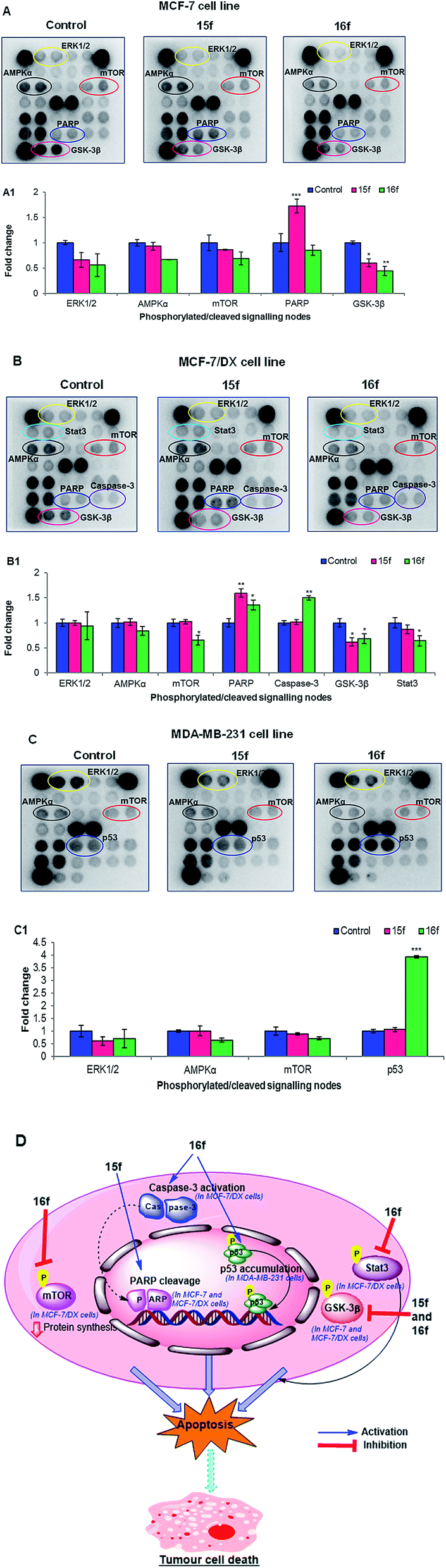 | ||
| Fig. 4 Chemiluminescent array images of the PathScan Intracellular Signalling array kit revealing various phosphorylated/cleaved signalling nodes in (A) MCF-7 cells, (B) MCF-7/DX cells and (C) MDA-MB-231 cells after the treatment with compounds 15f and 16f at 10 μM for 24 h. Cells without treatment serve as control. Bar chart representing the fold change in the integrated density of phosphorylated/cleaved signalling nodes in the array image of (A1) MCF-7 cells, (B1) MCF-7/DX cells and (C1) MDA-MB-231 cells after the treatment with and without compounds 15f and 16f at 10 μM for 24 h. Data are expressed as the mean ± standard error of the mean (SEM) (n = 4). Statistical significance was estimated with respect to the control by one-way ANOVA, followed by Bonferroni's post hoc test (*p < 0.05, **p < 0.01 and ***p < 0.001). (D) Molecular mechanism of action of 15f and 16f in MCF-7, MCF-7/DX and MDA-MB-231 cell lines. | ||
Interestingly, treatment of MDA-MB-231 cells with compound-16f showed a 2-fold increase in the levels of p53 compared to the control. MDA-MB-231 cells have a mutant p53 gene49,50 that is more stable and oncogenic unlike the wild type p53 in MCF-7 cells.51 The presence of mutant p53 drives the invasiveness of the tumour.52,53 However, the increase in the serine-15 phosphorylated p53 (mutant) in compound-16f treated MDA-MB-231 cells [than the control (untreated)] linked with its profound activity suggest that compound-16f induces Ser-15 phosphorylation of a mutant p53 in MDA-MB-231 cells, which probably leads to the restoration of wild-type p53 DNA binding activity and induction of cell death. A similar observation has been reported for resveratrol (a chalcone) wherein cell death was induced by restoring the wild-type p53 DNA binding activity via the phosphorylation of a mutant p53 at serine-15 in prostate cancer DU145 cells.54–56 This might explain the higher activity of compound-16f against MDA-MB-231 cells (IC50 = 8.9 μM for 16f vs. IC50 > 10 μM for 15f).
Overall, these results correlate well with the in vitro findings, and suggest that for the MCF-7 and MCF-7/DX cell lines, the anti-proliferative effects are mediated via ER-independent cleavage of PARP and downregulation of GSK-3β. For the MDA-MB-231 cell line, activity could be due to restoration of the wild-type p53 DNA binding activity of the mutant p53 tumour suppressor gene.
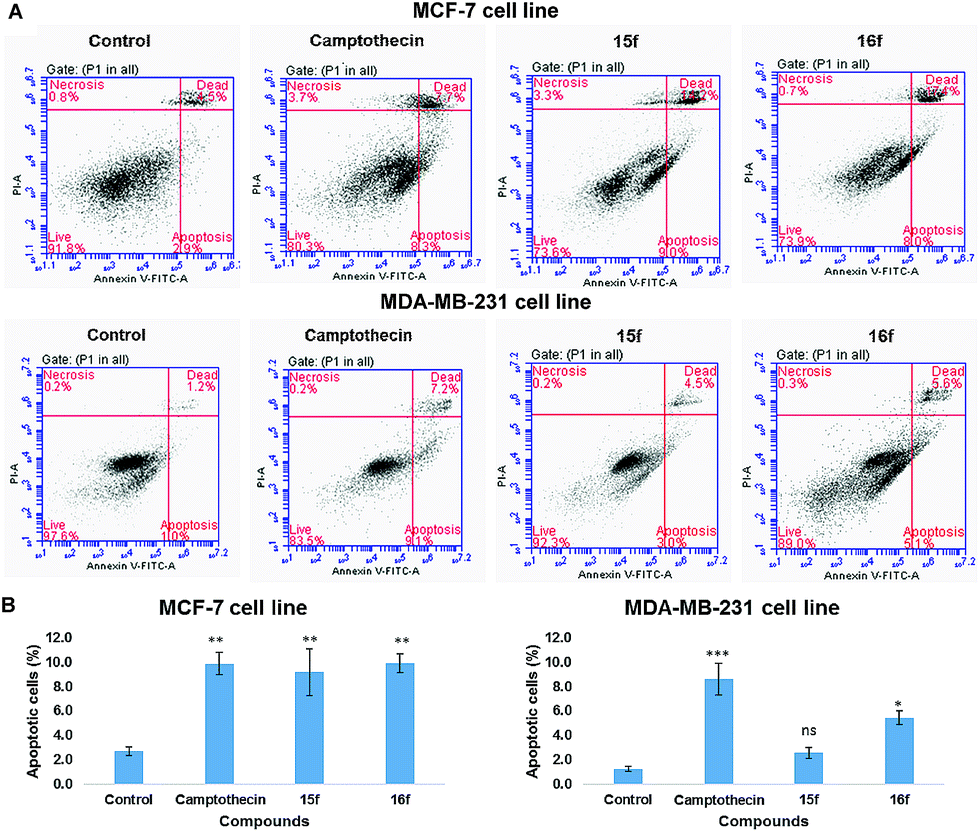 | ||
| Fig. 5 Apoptosis detection in MCF-7 and MDA-MB-231 cells by flow cytometry. MCF-7 cells were treated with compounds 15f and 16f at 1 μM and 5 μM respectively for 24 h and MDA-MB-231 cells were treated with compounds 15f and 16f at 10 μM and 9 μM respectively for 24 h. The quadrants were set based on the population of healthy, unstained cells in untreated samples (control) compared to cells treated with 5 μM camptothecin for 24 h. (A) Representative figures showing population of unstained live (annexin V− PI−, lower left), apoptotic (annexin V+ PI−, lower right), dead (annexin V+ PI+, upper right) and necrotic (annexin V− PI+, upper left) cells. (B) Bar chart represent the quantification of the apoptotic cells induced upon the treatment of MCF-7 and MDA-MB-231 cells with compounds 15f and 16f. Data are expressed as mean ± standard error of the mean (SEM), n = 3. Statistical significance was estimated with respect to the control (untreated sample) by one-way ANOVA, followed by Bonferroni's post hoc test (ns = nonsignificant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). | ||
| Panel/cell line | GI50 (μM) | |
|---|---|---|
| 15f | 16f | |
| a Note: sub-micromolar range GI50 values are highlighted in bold. | ||
| Leukaemia | ||
| CCRF-CEM | 4.13 | 2.53 |
| HL-60 | 2.99 | 1.66 |
| K-562 | 3.39 | 2.47 |
| MOLT-4 | 3.93 | 2.16 |
| RPMI-8226 | 2.95 | 1.32 |
| SR | 3.97 | 2.54 |
|
||
| Non-small cell lung cancer | ||
| A549/ATCC | 9.20 | 3.26 |
| EKVX | 17.3 | 17.9 |
| HOP-62 | 1.95 | 1.50 |
| HOP-92 | 1.76 | 1.69 |
| NCI-H226 | 14.6 | 7.22 |
| NCI-H23 | 1.95 | 1.52 |
| NCI-H322M | 2.23 | 1.89 |
| NCI-H460 | 3.09 | 0.97 |
| NCI-H522 | 2.57 | 1.16 |
|
||
| Colon cancer | ||
| COLO 205 | 16.8 | 10.7 |
| HCC-2998 | 1.74 | 2.28 |
| HCT-116 | 2.78 | 2.22 |
| HCT-15 | 0.48 | 0.85 |
| HT29 | 19.4 | 4.56 |
| KM12 | 2.17 | 3.35 |
| SW-620 | 3.85 | 2.60 |
|
||
| CNS cancer | ||
| SF-268 | 3.57 | 2.18 |
| SF-295 | 8.97 | 4.01 |
| SF-539 | 3.48 | 1.98 |
| SNB-19 | 3.85 | 2.95 |
| SNB-75 | 2.91 | 4.55 |
| U251 | 3.36 | 2.44 |
|
||
| Melanoma | ||
| LOX IMVI | 3.17 | 1.18 |
| MALME-3M | 1.81 | 1.87 |
| M14 | 3.25 | 2.01 |
| MDA-MB-435 | 3.46 | 2.98 |
| SK-MEL-2 | 2.69 | 2.27 |
| SK-MEL-28 | 4.67 | 2.94 |
| SK-MEL-5 | 2.05 | 1.79 |
| UACC-257 | 2.93 | 2.88 |
| UACC-62 | 3.27 | 2.09 |
|
||
| Ovarian cancer | ||
| IGROV1 | 1.14 | 2.56 |
| OVCAR-3 | 1.96 | 1.23 |
| OVCAR-4 | 0.57 | 2.03 |
| OVCAR-5 | 12.1 | 5.83 |
| OVCAR-8 | 3.34 | 2.28 |
| NCI/ADR-RES | 2.82 | 1.43 |
| SK-OV-3 | 3.61 | 2.60 |
|
||
| Renal cancer | ||
| 786-0 | 7.35 | 2.89 |
| A498 | 1.22 | 2.15 |
| ACHN | 2.53 | 2.35 |
| CAKI-1 | 3.12 | 2.65 |
| RXF393 | 4.22 | 2.51 |
| SN12C | 2.83 | 2.61 |
| TK-10 | 4.60 | 4.16 |
| UO-31 | 1.28 | 1.02 |
|
||
| Prostate cancer | ||
| PC-3 | 3.21 | 2.92 |
| DU-145 | 4.26 | 2.67 |
|
||
| Breast cancer | ||
| MCF-7 | 0.18 | 1.46 |
| MDA-MB-231/ATCC | 2.20 | 1.81 |
| HS 578T | 5.26 | 6.69 |
| BT-549 | 3.40 | 2.19 |
| T-47D | 0.03 | 1.27 |
| MDA-MB-468 | 0.47 | 2.08 |
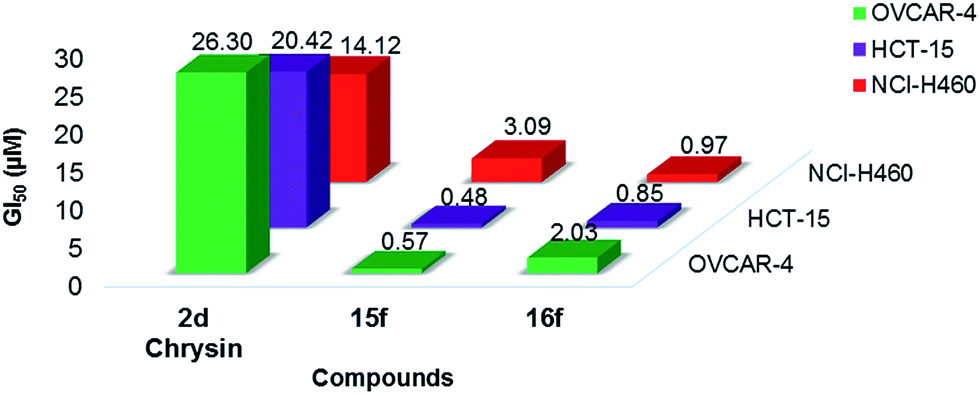 | ||
| Fig. 6 Comparison of GI50 values of compounds 15f and 16f with 2d (chrysin, a well-known natural flavone). Data for 2d (chrysin) were obtained from NCI/DTP screening: December 2010 (NSC407436). | ||
Conclusions
A library of 76 compounds containing structurally related methoxy and hydroxy flavones, and their 4-thio analogues, has been designed, synthesised and evaluated for anti-proliferative activity against the breast cancer cell lines MCF-7 (ER +ve), MCF-7/DX (ER +ve, anthracycline resistant) and MDA-MB-231 (ER −ve). Within this library, 42 compounds were prepared and characterised for the first time within our laboratory. The study provided significant insight into the structural features required for enhancing the anti-proliferative profiles of flavones, and identified two novel hydroxy 4-thioflavones 15f and 16f, as lead anti-proliferative agents (15f (IC50 = 1.0 ± 0.1 μM and 9 ± 0.1 μM against MCF-7 and MCF-7/DX cell lines, respectively) and 16f (IC50 = 4.9 ± 0.7 μM, 6.5 ± 0.4 μM and 8.9 ± 0.8 μM against MCF-7, MCF-7/DX and MDA-MB-231 cell lines, respectively)). A systematic SAR study highlighted the presence of free hydroxyl groups, and the B-ring phenyl groups, as essential for enhanced anti-proliferative activities. Replacement of the 4-CO functional group with the 4-CS functional group also enhanced the anti-proliferative activities. Incorporation of lipophilic electron withdrawing groups at C-4′ of the B-ring phenyl was found to be favourable, and the increased lipophilicity correlates well with the greater anti-proliferative activities. Molecular mechanistic studies have suggested that the anti-proliferative effects of flavones 15f and 16f are mediated via ER-independent cleavage of PARP and downregulation of GSK-3β in the case of MCF-7 and MCF-7/DX cell lines. For the MDA-MB-231 cell line, a probable restoration of the wild-type p53 DNA binding activity of mutant p53 tumour suppressor gene was indicated. Taken together, these results demonstrate that the novel thioflavones 15f and 16f offer considerable potential as anti-proliferative agents within medicinal chemistry programmes focussed on cancer. In addition, the SARs derived herein are likely to contribute significantly to the design and synthesis of novel thioflavones for further therapeutic applications.
Experimental
Materials and methods
:1 v/v) and these stocks were then appropriately diluted with the complete culture medium and, the ethanol and DMSO levels were maintained below 1% in the test concentrations. The IC50 values were calculated using MS excel 2010. Molecular docking was performed using SYBYL-X-2.1 software.IC50 values presented here correspond to the concentration (T/C) × 100 = 50, where T = absorbance of the test well after 72 h exposure to the test compound, C = absorbance of control well after 72 h.
GI50 values presented here involve a correction for the cell count at time zero. Therefore the GI50 value for a test drug corresponds to the concentration [(T − T0)/(C − T0)] × 100 = 50, where T = absorbance of the test well after 48 h exposure to the test compound, C = absorbance of control after 48 h and T0 = absorbance at time zero.
Molecular docking
64 suite. If the root mean square deviation (RMSD) value between the real and the best-scored conformations is equal to or less than 2.0 Å (representing the grid spacing used for the docking procedure), then the docking process was considered successful.65 In this case the docking procedure was first validated by re-docking the extracted co-crystallised ligand of human estrogen receptor-α (ERα) (2-phenyl-1-[4-(2-piperidin-1-yl-ethoxy)-phenyl]-1,2,3,4-tetrahydro-isoquinolin-6-ol) into the prepared target protein to be used for docking. The RMSD between the docked conformation, as generated by the docking algorithm and the native co-crystallised ligand conformation was found to be 0.12 Å, which was well within the 2 Å grid spacing used in the docking procedure, indicating that the docking method to be used was reliable and valid. Furthermore, the interactions between the docked ligand and the prepared target receptor mimicked those observed in the crystal structure of the same.The Surflex-X docking algorithm docks a given ligand to a receptor using a flexible ligand and a semi-flexible receptor; in this case the peptides were allowed to be fully flexible while the receptor was semi-flexible. This approach allows for optimisation of potentially favourable molecular interactions, such as those defined by hydrogen bond and van der Waal forces. The docking results yield a docking score, which takes into consideration entropic, polar, hydrophobic, repulsive and desolvation factors. Here, the docking scores were expressed in −log10(Kd) units to represent binding affinities, where Kd is the dissociation constant. The free energy of binding of the ligand to the protein was extrapolated from eqn (1).
|
Free energy of binding = RTlogeKd
| (1) |
The docking results were visualised using the programme PyMOL67,68 and the molecular interactions of the docked ligands were analysed by the programme CONTACTS, as provided in the CCP4 suite of programmes.64,69,70 Potential hydrogen bonds were assigned if the distance between two electronegative atoms was less than 3.3 Å, whereas any separation greater than 3.3 Å, but less than 4.5 Å, was considered a van der Waal interaction.
(a) Preparation of cell lysate. MCF-7 and MCF-7/DX cells (8 × 104 cell per mL) and MDA-MB-231 cells (4 × 104 cells per mL) were seeded into 24 well plates (2 mL per well) and incubated for 24 h. After 24 h, the cells were treated with the test compounds at 10 μM concentration for 24 h. Following this 24 h exposure, the cells were washed with ice-cold 1× phosphate-buffered saline and lysed in 1× cell lysis buffer provided (phosphotase and protease inhibitors added). These lysates were quantified using the BCA protein assay.
(b) Assay procedure. The array blocking buffer was added to each well of the glass slide provided and incubated for 15 min at room temperature. Subsequently, the cell lysate, diluted to 0.3 mg mL−1 in array diluents, was added to each well and incubated for 2 h at room temperature. Subsequent to washing, the detection antibody cocktail was added to each well and incubated for 1 h at room temperature. Horseradish peroxidase (HRP)-linked streptavidin was added to each well and incubated for 30 min at room temperature. The slide was then covered with LumiGLO/peroxide reagent (Cell Signaling Technology) and exposed to film for 2–30 s. The image was captured by a digital imaging system, ImageQuant LAS 4000 (GE Healthcare).
000 cells per sample was collected and dot plots of FL1 vs. FL3 were generated. The quadrants were set based on the population of healthy, unstained cells in untreated samples compared to cells treated with a known apoptotic inducer camptothecin (5 μM) for 24 h. BD CSampler™ software was used to calculate the percentage of the cells in the respective quadrants. The experiment was performed in triplicate.Conflict of interest
The authors declare no competing financial interest.Abbreviations
| DMSO | Dimethylsulfoxide |
| DCM | Dichloromethane |
| EtOAc | Ethylacetate |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NMR | Nuclear magnetic resonance |
| IR | Infrared |
| FTMS | Fourier transform mass spectroscopy |
| ESI | Electron spray ionisation |
| ERK1/2 | Extracellular signal-regulated kinase |
| mTOR | Mammalian target of rapamycin |
| PARP | Poly(ADP-ribose) polymerase |
| AMPKα | 5′-Adenosine monophosphate-activated protein kinase |
| p53 | Phosphoprotein53 |
| GSK-3β | Glycogen synthase kinase-3β |
| Bad | Bcl-2-associated death promoter |
| PRAS40 | Proline-rich Akt substrate of 40 kDa |
| PI3K | Phosphatidylinositol-4,5-bisphosphate-3-kinase |
| MMFF94 | Merck molecular force field 94 |
Acknowledgements
Financial support to DR from the Felix trust is gratefully acknowledged. We thank Amit Kumar Rajora for his help with the sample preparation for microarray experiments. We also thank the University of Reading for the provision of the Chemical Analysis Facility and Dr Philip Dash, University of Reading, UK for providing MDA-MB-231 cells. We gratefully acknowledge NCI, USA for accepting and screening our compounds against 60 cell lines [NSC reference numbers – 783090 (15f) and 783091 (16f)].References
- A. L. Harvey, R. Edrada-Ebel and R. J. Quinn, Nat. Rev. Drug Discovery, 2015, 14, 111–129 CrossRef CAS PubMed
.
- L. Ouyang, Y. Luo, M. Tian, S.-Y. Zhang, R. Lu, J.-H. Wang, R. Kasimu and X. Li, Cell Proliferation, 2014, 47, 506–515 CrossRef CAS PubMed
- W. Ren, Z. Qiao, H. Wang, L. Zhu and L. Zhang, Med. Res. Rev., 2003, 23, 519–534 CrossRef CAS PubMed
- P. Batra and A. K. Sharma, 3 Biotech, 2013, 3, 439–459 CrossRef
- J. Carini, F. Klamt and V. Linck, RSC Adv., 2014, 4, 3131–3144 RSC
- A. K. Verma and R. Pratap, Nat. Prod. Rep., 2010, 27, 1571–1593 RSC
- R. S. Keri, S. Budagumpi, R. K. Pai and R. G. Balakrishna, Eur. J. Med. Chem., 2014, 78, 340–374 CrossRef CAS PubMed
- A. Gaspar, M. J. Matos, J. Garrido, E. Uriarte and F. Borges, Chem. Rev., 2014, 114, 4960–4992 CrossRef CAS PubMed
- C. Kanadaswami, L. T. Lee, P. P. H. Lee, J. J. Hwang, F. C. Ke, Y. T. Huang and M. T. Lee, In Vivo, 2005, 19, 895–910 CAS
- D. Ravishankar, A. K. Rajora, F. Greco and H. M. I. Osborn, Int. J. Biochem. Cell Biol., 2013, 45, 2821–2831 CrossRef CAS PubMed
- S. Kawaii, Y. Tomono, E. Katase, K. Ogawa and M. Yano, Biosci., Biotechnol., Biochem., 1999, 63, 896–899 CrossRef CAS PubMed
- A. M. Senderowicz, Invest. New Drugs, 1999, 17, 313–320 CrossRef CAS PubMed
- S. Burdette-Radoux, R. G. Tozer, R. C. Lohmann, I. Quirt, D. S. Ernst, W. Walsh, N. Wainman, A. D. Colevas and E. A. Eisenhauer, Invest. New Drugs, 2004, 22, 315–322 CrossRef CAS PubMed
- J. A. Jones, A. S. Rupert, M. Poi, M. A. Phelps, L. Andritsos, R. Baiocchi, D. M. Benson, K. A. Blum, B. Christian, J. Flynn, S. Penza, P. Porcu, M. R. Grever and J. C. Byrd, Am. J. Hematol., 2014, 89, 19–24 CrossRef CAS PubMed
- C. Loguercio and D. Festi, World J. Gastroenterol., 2011, 17, 2288–2301 CrossRef CAS PubMed
- D. Ferry, A. Smith and J. Malkhandi, Clin. Cancer Res., 1996, 659–668 CAS
- P. J. Mulholland, D. R. Ferry, D. Anderson, S. A. Hussain, A. M. Young, J. E. Cook, E. Hodgkin, L. W. Seymour and D. J. Kerr, Ann. Oncol., 2001, 12, 245–248 CrossRef CAS PubMed
- S. Kawa II, Y. Tomono, E. Katase, K. Ogawa and M. Yano, Biosci., Biotechnol., Biochem., 2014, 63, 896–899 CrossRef PubMed
- J. Yáñez, V. Vicente, M. Alcaraz, J. Castillo, O. Benavente-García, M. Canteras and J. A. L. Teruel, Nutr. Cancer, 2004, 49, 191–199 CrossRef PubMed
- J. A. Manthey and N. Guthrie, J. Agric. Food Chem., 2002, 50, 5837–5843 CrossRef CAS PubMed
- P. Parajuli, N. Joshee, A. M. Rimando, S. Mittal and A. K. Yadav, Planta Med., 2009, 75, 41–48 CrossRef CAS PubMed
- M. Lopez-Lazaro and M. Akiyama, Curr. Med. Chem.: Anti-Cancer Agents, 2002, 2, 691–714 CrossRef CAS PubMed
- M. López-Lázaro, M. Gálvez, C. Martín-Cordero and M. J. Ayuso, Stud. Nat. Prod. Chem., 2002, 27, 891–932 Search PubMed
- J. F. W. McOmie, M. L. Watts and D. E. West, Tetrahedron, 1968, 24, 2289–2292 CrossRef CAS
- W. Baker, J. Chem. Soc., 1933, 1381–1389 RSC
- M. P. Cava and M. I. Levinson, Tetrahedron, 1985, 41, 5061–5087 CrossRef CAS
- J. Allan and R. Robinson, J. Chem. Soc., 1926, 129, 2334–2336 RSC
- D. Ravishankar, K. A. Watson, S. Y. Boateng, R. J. Green, F. Greco and H. M. I. Osborn, Eur. J. Med. Chem., 2015, 97, 259–274 CrossRef CAS PubMed
- I. L. Martins, C. Charneira, V. Gandin, J. L. Ferreira da Silva, G. C. Justino, J. P. Telo, A. J. S. C. Vieira, C. Marzano and A. M. M. Antunes, J. Med. Chem., 2015, 58, 4250–4265 CrossRef CAS PubMed
- J. G. Topliss, J. Med. Chem., 1972, 15, 1006–1011 CrossRef CAS PubMed
- P. F. Devitt, A. Timoney and M. A. Vickars, J. Org. Chem., 1961, 26, 4941–4944 CrossRef CAS
- C. Pouget, F. Lauthier, A. Simon, C. Fagnere, J. P. Basly, C. Delage and A. J. Chulia, Bioorg. Med. Chem. Lett., 2001, 11, 3095–3097 CrossRef CAS PubMed
- N. Yao, C. Y. Chen, C. Y. Wu, K. Motonishi, H. J. Kung and K. S. Lam, J. Med. Chem., 2011, 54, 4339–4349 CrossRef CAS PubMed
- K. Sak, Pharmacogn. Rev., 2014, 8, 122–146 CrossRef CAS PubMed
- M. A. Indap, R. Susarla, L. Motiwale and K. V. K. Rao, Indian J. Pharm. Sci., 2006, 68, 465–469 CrossRef CAS
- S. Yadegarynia, A. Pham, A. Ng, D. Nguyen, T. Lialiutska, A. Bortolazzo, V. Sivryuk, M. Bremer and J. B. White, Nat. Prod. Commun., 2012, 7, 1295–1304 CAS
- E. Mechetner, A. Kyshtoobayeva, S. Zonis, H. Kim, R. Stroup, R. Garcia, R. J. Parker and J. P. Fruehauf, Clin. Cancer Res., 1998, 4, 389–398 CAS
- J. B. Daskiewicz, F. Depeint, L. Viornery, C. Bayet, G. Comte-Sarrazin, G. Comte, J. M. Gee, I. T. Johnson, K. Ndjoko, K. Hostettmann and D. Barron, J. Med. Chem., 2005, 48, 2790–2804 CrossRef CAS PubMed
- I. N. Sergeev, C.-T. Ho, S. Li, J. Colby and S. Dushenkov, Mol. Nutr. Food Res., 2007, 51, 1478–1484 CAS
- T. Walle, Int. J. Mol. Sci., 2009, 10, 5002–5019 CrossRef CAS PubMed
- T. Hirano, M. Gotoh and K. Oka, Life Sci., 1994, 55, 1061–1069 CrossRef CAS PubMed
- M. Lacroix and G. Leclercq, Breast Cancer Res. Treat., 2004, 83, 249–289 CrossRef CAS PubMed
- N. Sharma, M. Dobhal, Y. Joshi and M. Chahar, Pharmacogn. Rev., 2011, 5, 1 CrossRef PubMed
- S. Kumar and A. K. Pandey, Sci. World J., 2013, 2013 Search PubMed
- J. Renaud, S. F. Bischoff, T. Buhl, P. Floersheim, B. Fournier, C. Halleux, J. Kallen, H. Keller, J.-M. Schlaeppi and W. Stark, J. Med. Chem., 2003, 46, 2945–2957 CrossRef CAS PubMed
- T. Akama, Y. Shida, T. Sugaya, H. Ishida, K. Gomi and M. Kasai, J. Med. Chem., 1996, 39, 3461–3469 CrossRef CAS PubMed
- G. V. Chaitanya, A. J. Steven and P. P. Babu, Cell Commun. Signaling, 2010, 8, 31 CrossRef CAS PubMed
- L. M. Mooney, K. A. Al-Sakkaf, B. L. Brown and P. R. M. Dobson, Br. J. Cancer, 2002, 87, 909–917 CrossRef CAS PubMed
- M. Olivier, R. Eeles, M. Hollstein, M. A. Khan, C. C. Harris and P. Hainaut, Hum. Mutat., 2002, 19, 607–614 CrossRef CAS PubMed
- L. Hui, Y. Zheng, Y. Yan, J. Bargonetti and D. A. Foster, Oncogene, 2006, 25, 7305–7310 CrossRef CAS PubMed
- A. L. Gartel, C. Feliciano and A. L. Tyner, Oncol. Res., 2003, 13, 405–408 CrossRef PubMed
- M. Hsiao, J. Low, E. Dorn, D. Ku, P. Pattengale, J. Yeargin and M. Haas, Am. J. Pathol., 1994, 145, 702–714 CAS
- P. A. J. Muller, P. T. Caswell, B. Doyle, M. P. Iwanicki, E. H. Tan, S. Karim, N. Lukashchuk, D. A. Gillespie, R. L. Ludwig, P. Gosselin, A. Cromer, J. S. Brugge, O. J. Sansom, J. C. Norman and K. H. Vousden, Cell, 2009, 139, 1327–1341 CrossRef PubMed
- H.-Y. Lin, A. Shih, F. B. Davis, H.-Y. Tang, L. J. Martino, J. A. Bennett and P. J. Davis, J. Urol., 2002, 168, 748–755 CrossRef CAS PubMed
- H. Y. Lin, H. Y. Tang, F. B. Davis and P. J. Davis, Ann. N. Y. Acad. Sci., 2011, 1215, 79–88 CrossRef CAS PubMed
- A. Shih, S. Zhang, H. J. Cao, S. Boswell, Y.-H. Wu, H.-Y. Tang, M. R. Lennartz, F. B. Davis, P. J. Davis and H.-Y. Lin, Mol. Cancer Ther., 2004, 3, 1355–1364 CAS
- M. C. Willingham, J. Histochem. Cytochem., 1999, 47, 1101–1110 CrossRef CAS PubMed
- K. Emoto, N. Toyama-Sorimachi, H. Karasuyama, K. Inoue and M. Umeda, Exp. Cell Res., 1997, 232, 430–434 CrossRef CAS PubMed
- R. H. Shoemaker, Nat. Rev. Cancer, 2006, 6, 813–823 CrossRef CAS PubMed
- V. Vichai and K. Kirtikara, Nat. Protoc., 2006, 1, 1112–1116 CrossRef CAS PubMed
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed
- A. N. Jain, J. Med. Chem., 2003, 46, 499–511 CrossRef CAS PubMed
- A. N. Jain, J. Comput.-Aided Mol. Des., 2007, 21, 281–306 CrossRef CAS PubMed
- Collaborative Computational Project, Number 4, The CCP4 Suite: Programs for Protein Crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1994, 50, 760–763 Search PubMed.
- R. Wang, Y. Lu and S. Wang, J. Med. Chem., 2003, 46, 2287–2303 CrossRef CAS PubMed
- M. J. D. Powell, Math. Program., 1977, 12, 241–254 CrossRef
- W. DeLano, CCP4 Newsl, Protein Crystallography, 40, 2002.
- D. Seeliger and B. L. De Groot, J. Comput.-Aided Mol. Des., 2010, 24, 417–422 CrossRef CAS PubMed
- K. Cowtan, P. Emsley and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 233–234 CrossRef CAS PubMed
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: This includes the synthesis and characterisation of compounds 1a–f to 19a–f as well as figures summarising the anti-proliferative activities of compounds 1c–f to 5c–f against the MDA-MB-231 cell line and the anti-proliferative activities of compounds 6c–f to 11c–f against (A) MCF-7 cell line (B) MCF-7/DX cell line (C) MDA-MB-231 cell line. Dose response curves are also provided. See DOI: 10.1039/c6ra11041j |
| This journal is © The Royal Society of Chemistry 2016 |