
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Broadband-sensitive Ni2+–Er3+ based upconverters for crystalline silicon solar cells†
Hom N.
Luitel
*,
Shintaro
Mizuno
,
Toshihiko
Tani
and
Yasuhiko
Takeda
Toyota Central Research and Development Laboratories Inc., 41-1, Yokomichi, Nagakute, Aichi 480-1192, Japan. E-mail: e1698@mosk.tytlabs.co.jp; takeda@mosk.tytlabs.co.jp; Tel: +81 956 717134
First published on 27th May 2016
Abstract
We have developed Ni2+, Er3+ codoped CaZrO3 broadband-sensitive upconverters that significantly broaden the sensitive range, and hence overcome the shortcomings of conventional Er3+ doped upconverters used for crystalline silicon (c-Si) solar cells that utilize only a small fraction of the solar spectrum around 1550 nm. We have designed the combination of sensitizers and host material to utilize photons that are not absorbed by c-Si itself or Er3+ ions. Six coordinated Ni2+ ions substituted at the Zr4+ sites absorb (1060–1450) nm photons and transfer the energies to the Er3+ ions, and the Er3+ upconverts at 980 nm. Co-doping with monovalent charge compensators such as Li+ for high Er3+ solubilisation at the Ca2+ sites and multivalent ions (Nb5+) for stabilization of Ni2+ at the Zr4+ sites is essential. In addition to 1450–1600 nm (≈2 × 1020 m−2 s−1) photons directly absorbed by the Er3+ ions, we have demonstrated upconversion of 1060–1450 nm (≈6 × 1020 m−2 s−1) photons in the Ni2+ absorption band to 980 nm photons using the CaZrO3:Ni2+,Er3+ upconverters. Compared with the current density gain of present c-Si solar cells (∼40 mA cm−2), the upconverted photons could increase this by ∼7.3 mA cm−2, which is about 18% improvement. This architecture for broadband-sensitive upconversion may pave a new direction for the improvement in efficiency of the present c-Si solar cells to surpass the limiting conversion efficiency of single-junction solar cells.
1. Introduction
Incident solar radiation is energetically broad and poses challenges for efficient conversion to electricity using solar cells consisting of finite bandgap semiconductors.1 Even with an optimized crystalline silicon (c-Si) semiconductor, ∼30% of the incident radiation is not absorbed and is simply transmitted through the present c-Si solar cells. Considering the maximum conversion efficiency of single-junction solar cells, ∼33% sunlight into electricity in the radiative limit,2 the unused near-infrared (NIR) radiation provides an important opportunity for enhancing the efficiency of solar cells. Although the NIR photons have insufficient energy, combining two or more infrared photons to generate a single visible or ultraviolet photon, a process known as upconversion, enables these NIR photons to contribute to solar energy conversion.3,4 An upconversion layer can be placed at the back of a solar cell and converts a part of the transmitted photons to wavelengths that can be absorbed; it is straightforward to demonstrate a positive contribution from the upconversion layer.Upconversion in various materials in which rare-earth ions are doped have been reported.5–9 Among them, upconverters using Er3+ ions that convert 1550 nm photons to 980 nm photons are applied to c-Si solar cells,10–12 while those using a combination of Er3+ and Yb3+, which converts 970 nm to the visible photons, to amorphous silicon (a-Si), organic, and dye-sensitized solar cells.13–17 However, the absorption bands of rare-earth ions originating from f–f transitions in the NIR region (700–2100 nm) are narrow, and hence only a small fraction of the solar spectrum can be utilized.17–19 Thus, to realize highly efficient solar cells, efficient upconverters with sufficiently wide absorption spectra are desired. One of the approaches is to use a combination of rare earth ions with sensitizers that absorb photons in a broadband range, which are not directly absorbed by the rare-earth ions, and transfer the absorbed energies to the rare-earth ions. There are reports on upconversion of 970 nm photons to the visible (550, 650 nm) using the Er3+–Yb3+ pairs where organic dyes function as the sensitizers that absorb photons in a range of 700–850 nm and transfer the energies to the Yb3+ (ref. 19) and similarly Cr3+ for 600–650 nm.20 On the other hand, upconversion with a rather broadband sensitivity and a high efficiency has been demonstrated using triplet–triplet annihilation of organic molecules but the absorption range is limited up to 800 nm at present and thus can have applications only for a-Si, organic, and dye-sensitized solar cells.21–23
2. Materials design
There are detailed reports on absorption spectra of transition-metal ions that extend up to the NIR range.24–31 It has been demonstrated that six coordinated Ni2+ ions located at the centers of octahedrons have substantial absorption around 1100–1400 nm that can further be tuned in a wide range by manipulating the crystal field strength around the Ni2+ ions.26,30,32 Accordingly, its NIR emission band is also wide ranging from 1200 nm to longer than 1700 nm with extremely high quantum yield reaching unity in some crystals such as LiGa5O8.33 There are other reports of the Ni2+ emission bands in the NIR ranges25–27,34–37 that sufficiently overlap with the Er3+ absorption bands making possibility of Ni2+ → Er3+ resonance energy transfer (sensitization).32,38,39 Zhang et al. has reported that Ni2+ to Er3+ energy transfer or vice versa in glass ceramics depending on the host matrix and Ni2+ to Er3+ physical separation to achieve NIR broadband Stokes emission for amplifier and lasers. Furthermore, a high concentration of the Ni2+ sensitizers is required, because it is equivalent to high excitation intensity for the Er3+ emitters. In the similar way, the optimum Er3+ concentration should also be high, 10–25% range, to promote energy transfer between two neighbouring excited Er3+ ions in addition to excited state absorption in a single Er3+ ion.34Thus, the host materials to realize broadband-sensitive upconversion using Er3+ and Ni2+ dopants are selected according to the following three criteria: (1) the host material must include cations located at octahedron centers, and should not include tetrahedron or other symmetry sites that can be occupied by Ni2+ ions. Four co-ordinated Ni2+ ions located at tetrahedron centers and Ni3+ have lower energy levels26,30 and hence rapid relaxation occur rather than energy transfer to the Er3+ emitters after photoexcitation. (2) The ionic radius of the cations located at the octahedron centers should be close to or at least not much smaller than that of Ni2+ (0.069 nm) to enable a high Ni2+ concentration, otherwise introduced Ni2+ ions would change to smaller Ni3+ ions (0.056 nm) or segregate. (3) The host material should include bigger ions (favourably isovalent to Er3+) such that sufficient amount of Er3+ can be solubilised. Most widely used upconversion materials such as NaYF4:Er3+, Y2O2S:Er3+, or YF3:Er3+ (ref. 10 and 40) do not contain octahedron centers for Ni2+ hosting thus not suitable for the present purpose. We have already found that ABO3 type perovskite structures such as the LaGaO3 structure fulfil these criteria, because Ga ions (B sites) are located at the octahedron centers and the ionic radius of Ga3+ (0.062 nm) is similar to that of Ni2+. La3+ ions (A sites) are isovalent to Er3+ ions with very similar ionic radii. Under such materials design, we have successfully demonstrated the broadband-sensitive upconversion utilizing the Ni–Er pairs in La(Ga,Sc)O3, where the Ni2+ sensitizers absorb the photons around 1100–1400 nm and transfer the absorbed energies to the Er3+ ions, followed by Er3+ upconversion at around 980 nm, highly suitable for c-Si solar cells.41 However, the Ni-sensitized Er-upconverter exhibited low efficiency, and La and Sc used in the La(Ga,Sc)O3 compounds are extremely expensive. For practical applications, inexpensive materials with high upconversion efficiency are desired.
It has been reported that alkaline earth perovskites such as CaZrO3 are good hosts for rare earth doped phosphors and can solubilize as high as 20% rare earth ions when proper charge compensators are utilized.42 The crystal structure of CaZrO3 consists of ZrO6 octahedra and CaO12 polyhedra as depicted in Fig. 1(c). The 6-coordinated Zr4+ ions occupy the centres of octahedra. The ionic radius of Zr4+ is 0.072 nm (6-coordination) while that of Ca2+ is 0.112 nm (12-coordination).43 Thus, it is expected that codoped Ni2+ ions (0.069 nm) occupy Zr4+ sites while Er3+ ions (0.106 nm) occupy the Ca2+ sites. When the tetravalent Zr4+ ions are substituted with divalent Ni2+ ions, oxygen vacancies are created for charge neutrality, which would cause parasitic absorption and nonradiative relaxation of excited Ni2+ and Er3+ ions. Further, Ni2+ ions located at the higher valence sites (Zr4+) would change to Ni3+ ions and do not contribute to upconversion as described above.30 Nb5+ ions seem suitable as the charge compensators because they are stable and the ionic radius (Nb5+ ≈ 0.064 nm) is close to that of Zr4+ (0.072 nm). Furthermore, it has been reported that the substitution of larger or smaller ions into host cation sites remarkably distorts the crystal field from the ideal symmetry and hence changes the crystal field splitting.44 As a result, the f–f transition probabilities improve leading to increased absorption and upconversion. In the CaZrO3:Er system, substitution of a part of the Ca2+ ions with smaller Er3+ ions distorts the crystal structure and gives the possibility of improved upconversion. In addition, substitution of smaller (Li+) or bigger (K+) ions for the Ca2+ ions further distorts the CaZrO3 crystal, which can more remarkably increase the upconversion efficiency (Table 1).
 | ||
| Fig. 1 (a) XRD patterns of CaZrO3 doped with 0.2 mol% Ni and different mol% Er. For comparison, standard pdf data of CaZrO3 (red solid lines) and ErZrO3.5 (blue dotted lines) are also shown. (b) SEM image of the typical sample and (c) crystal structure of CaZrO3 created by VESTA program. | ||
| Metal ions | Er3+ | Ca2+ | Li+ | Na+ | K+ |
| Ionic radii (nm) (12 coordination) | 0.106 | 0.112 | 0.092 | 0.118 | 0.151 |
| Metal ions | Ni2+ | Ni3+ | Zr4+ | Nb5+ | |
| Ionic radii (nm) (6 coordination) | 0.069 | 0.056 | 0.072 | 0.064 |
Therefore, in the present study we have elaborately investigated the broadband-sensitive upconversion characteristics in the newly designed Ni2+–Er3+ codoped CaZrO3 that can significantly improve the conversion efficiency of present c-Si solar cells.
3. Experiments
3.1. Synthesis of Er, Ni-codoped samples
We synthesized powder samples of CaZrO3 codoped with Er3+ and Ni2+. Equivalent amount of Li+ ions (or Na+/K+) to that of Er3+ ions and double amount of Nb5+ ions to that of Ni2+ ions were also co-doped for charge balance unless specified. Because Er3+ occupy Ca2+ sites and Ni2+, Nb5+ substitute Zr4+ sites, the compositions were determined as (ErxLixCa1−2x)(Zr1−3yNiyNb2y)O3. The compound oxide powders were synthesized using component metal-oxide or carbonate powders by the solid state reaction method. Predetermined amounts of the oxides/carbonates (Kojundo Kagaku, Japan) were well mixed with the help of small amount of ethanol and then dried at RT. Next the dry powders were again mixed well and heat-treated at 1400 °C for 6 h in air for reaction and crystallization. As synthesized powders might contain mixture of Ni2+ and Ni3+, post annealing of the powder samples was carried out at 800 °C in N2 gas flow to convert all the Ni3+ ions to Ni2+ ions.The crystalline structures were identified by X-ray diffraction (XRD) using Cu-Kα line in a range from 10 to 80 degree. Scanning electron microscope (SEM) observations were carried out using Hitachi SU3500.
3.2. Optical measurements
Each powder sample was sandwiched between two glass plates with a 0.5 mm thick spacer. Absorption spectra were measured using an integrating sphere. To determine the photon flux at the Ni2+ or Er3+ absorption bands, we have integrated the standard AM1.5 spectral solar irradiation at earth surface45 in the absorption range of Ni2+ and Er3+, respectively. The reference spectrum is available in irradiance (W m−2 nm−1) form, that was converted to the photon flux and current density forms. Continuous wave (CW) laser diodes of ∼1 mW output power were used as excitation sources to acquire upconversion and Stokes emission spectra. Excitation beam intensity (mW cm−2) at the sample surface was obtained using a power meter and a knife-edge and it was in the order of 106 mW cm−2. An optical parametric oscillator (OPO) pumped by the third harmonic of a Nd-YAG laser (7 ns pulse duration, 5–20 mW output power depending on output wavelengths) was employed for evaluation of wavelength dependent upconversion sensitivity and time-resolved measurements. Appropriate bandpass filters were used to receive emitted photons of desired wavelengths. Si and InGaAs photodiodes were used to detect the 980 nm and longer wavelength photons, respectively, and the output signal was accumulated using a storage oscilloscope.4. Results and discussion
Fig. 1(a) shows XRD patterns of the CaZrO3 doped with different mol% Er. The measured diffraction peaks of the powder samples matched well to those of the standard CaZrO3 card data (PDF# 01-080-6213), and confirmed that the samples were well crystallized in the orthorhombic phase (oP20). The sharp peaks are indication of good crystallization of the particles. The SEM image presented in Fig. 1(b) with well determined facets further confirmed the highly crystallized particles. The particles dimension varied over a range of 1–5 μm. The lattice constants calculated from the XRD result of the undoped CaZrO3 sample are a = 5.5861 Å, b = 8.012 Å and c = 5.7568 Å, which is in good agreement with the standard JCPDS card data (PDF# 01-080-6213). With increasing Er-doping concentration, the lattice constants (a, b, c) and cell volume gradually decreased as summarised in the ESI S1† indicating smaller Er3+ (0.106 nm) ions occupied the larger Ca2+ (0.112 nm) sites. However, at/above 15 mol% Er addition, no further lattice contraction was observed indicating the limiting solid solubility of the Er3+ ions in the CaZrO3 host, which was confirmed by existence of distinct XRD peaks of ErZrO3.5 phase (≈2θ = 30 degree) in the XRD diagram in Fig. 1(a). On the other hand, no distinct lattice changes were observed by the Ni substitution at the Zr sites because the amount of Ni was very small (<0.5 mol%) in addition to the fact that the ionic size of Ni2+ (0.069 nm) is very similar to that of Zr4+ (0.072 nm). Further, because trivalent Er3+ substituted divalent Ca2+ ions, charge unbalance limited the solubility of Er3+ in the CaZrO3 matrix as observed by the existence of Er2O3 peaks (2θ = 29.4 degree; PDF# 01-073-6274) in the Li free sample as shown in Fig. S2.† When equal amount of monovalent charge compensators such as Li+, Na+, K+ were codoped together with Er3+, the solid solubility of Er3+ in the CaZrO3 host increased, and as a result the Er2O3 phase disappeared (see ESI Fig. S2†). However, lower formation energy of pyrochlore structures (−3.88 eV) compared to the perovskite structures, CaZrO3 (−3.66 eV), the ErZrO3.5 pyrochlore phase was unavoidable at higher Er-doping concentration.46 Reflecting the fact that codoped Li+ (Na+/K+) occupied Ca2+ sites, the lattice parameters and cell volume changed according to the ionic radii of these alkali ions (Fig. S2†).The absorption spectra of the CaZrO3 doped with 10 mol% Er and different mol% of Ni are presented in Fig. 2 and S3 (refer ESI S3† for visible absorption range). Sharp absorption peaks located at 970, 800, 660, 520, 490, 410, 380 nm and comparatively broad absorption band around (1450–1600) nm were assigned to the f–f transitions of Er3+ ions,24,34 while rather broad but comparatively weak absorption bands located around (1050–1500) nm, (680–860) nm and (390–460) nm were originated from the 3A2(3F) to 3T2(3F), 3T1(3F) and 3T1(3P) optically allowed transitions, respectively, of six coordinated Ni2+ ions.20,32 No distinct Ni3+ absorption bands were identified indicating that most of the doped Ni stabilised as Ni2+ ions.25,33 We used Nb5+ ions as charge compensator in double amount to that of doped Ni that might have stabilized the added Ni as Ni2+ ions. The relative locations of the energy levels of the Er3+ and Ni2+ ions are illustrated in Fig. 2(b). Furthermore, the Ni2+ absorption bands remarkably increased with increasing doped Ni2+ ion concentration. The extra broad Ni2+ absorption band (1060–1500 nm) in combination with the Er3+ absorption band (1450–1600 nm) covers the most of the NIR solar spectrum (1050–1600 nm) which is not absorbed by c-Si solar cells themselves and will have a great advantage for the spectral conversion purpose.
 | ||
| Fig. 2 (a) Absorption spectra of the CaZrO3:10 mol% Er codoped with different mol% of Ni exhibiting Er–Ni combined broadband absorption and (b) energy level diagram of the Ni2+ and Er3+ ions in the CaZrO3 host with possible transitions and Ni → Er energy transfer. | ||
Fig. 3(a) and (b) show the upconversion spectra of the various mol% of Er (Li) doped CaZrO3:0.2 mol% Ni (Nb) samples when excited at 1490 and 1300 nm, respectively. Under the 1490 nm excitation, corresponding to direct Er3+ excitation, clear upconverted emissions at around 980 nm were observed (Fig. 3(a)). In addition, very similar upconverted emissions appeared while excited at 1300 and 1180 nm (for 1180 nm excited UC see Fig. S5†) despite the excitation light was not absorbed by the Er3+ ions themselves. The maximal upconversion intensity was achieved at 15 mol% Er doping concentration under both the 1300 and 1490 nm excitations. Further, it has been confirmed that the upconverted emission at 980 nm was proportional to the square of the Er concentrations for both excitation wavelengths at lower concentrations as depicted in the insets of Fig. 3(a) and (b). The power dependent upconversion intensity measurements confirmed that the present upconversion is two photon processes under both the 1490 and 1300 nm excitations (Fig. 3(c)).
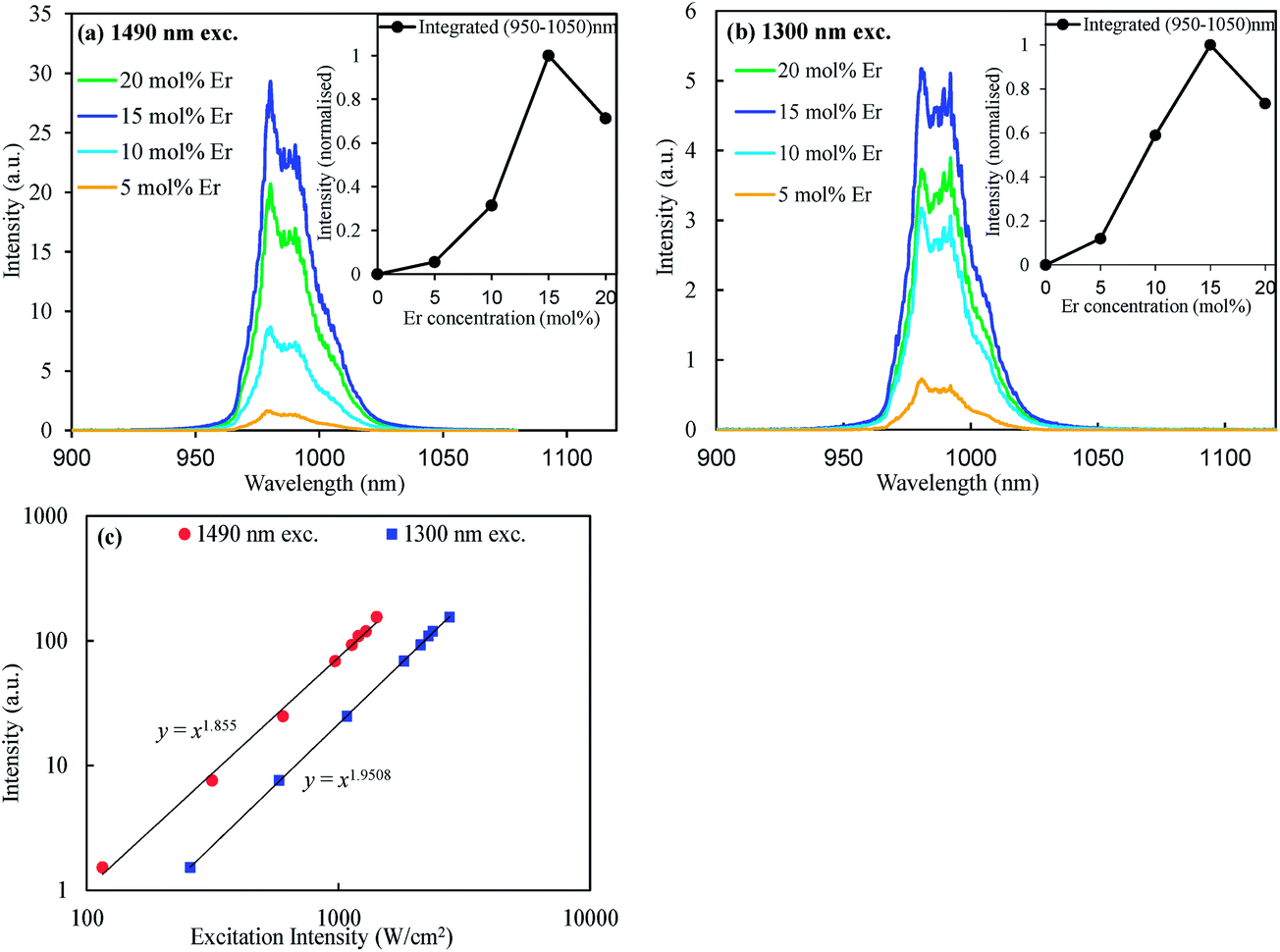 | ||
| Fig. 3 Upconversion spectra of the CaZrO3:0.2 mol% Ni codoped with different mol% of Er excited at (a) 1490 nm, and (b) 1300 nm. (c) Power-dependent UC intensity of the CaZrO3:0.2 mol% Ni2+,15 mol% Er3+ under 1490 and 1300 nm excitations. Inset in the (a) and (b) are the dependence of the UC intensities integrated over 950–1050 nm with the Er3+ ion concentration. | ||
Fig. 4 compares the Stokes emissions of the Ni (Nb) only doped and Er, Ni (Li, Nb)-codoped samples excited at 1180 nm. For the Ni only doped (no Er) sample, broad emission band ranging from 1300 nm to 1600 nm originated from the 3T2(3F) → 3A2(3F) transition of Ni2+ was observed.26–33 However, by introducing Er3+ (Li+) co-dopants, the Ni2+ emission almost disappeared and Er3+ emissions at around 1550 nm (4I13/2 → 4I15/2) and 980 nm (4I11/2 → 4I15/2) appeared (see Fig. S5† for 1180 nm excited UC at 980 nm). This suggests that the energies absorbed by the Ni2+ ions transferred to the Er3+ ions, followed by the Er3+ emissions i.e. the Ni2+ ions act as donors while Er3+ acceptors. Further, under sufficient Er3+ co-doping (15 mol% or higher), the Ni2+ emission was almost perfectly quenched and only the Er3+ emission appeared, indicating the Ni → Er energy transfer efficiency was close to unity. The energy transfer efficiency estimated from the ratio of the Ni2+ emission intensities integrated over 1300–1450 nm in the Ni only doped sample (0.2 mol% Ni) and Ni–Er codoped sample (0.2 mol% Ni, 10 mol% Er) was as high as 84%. However, at lower Er3+ concentrations, the Er3+ ions were far apart from the Ni2+ ions and the Ni → Er energy transfer was less efficient, as a result, Ni2+ emission was detected although it was weak. The energy transfer efficiency and subsequent excited Er3+ number should increase with the Er3+ concentration, and consequently the Er3+ Stokes emission intensity. In fact, the Er3+ Stokes emission, which is a one-photon process, was proportional to the Er concentration (see Fig. S4†). On the other hand, Er3+ upconversion (980 nm) is a two-photon process (Fig. 3(c)); it means two excited Er3+ ions are utilised to emit a single upconverted photon at a shorter wavelength. Thus, the upconversion emission intensity should be proportional to the square of the Er3+ ion concentration, which was obeyed at relatively lower Er3+ concentrations. However, at higher Er3+-doping concentrations, the Ni → Er energy transfer efficiency saturated and concentration quenching processes predominates, consequently the upconversion and Stokes emissions weakened.34
 | ||
| Fig. 4 Stokes emission spectra of the Ni (Nb) only doped (dotted line) and different mol% of Er (Li) codoped CaZrO3 samples excited at 1180 nm. | ||
Effect of the Ni2+-doping concentration on the upconversion spectra of CaZrO3:15 mol% Er(Li) are presented in Fig. 5. At the beginning, the upconversion intensity increased with increasing Ni-doping concentration that is due to increased Ni absorption as depicted in Fig. 2(a). However, at slightly higher Ni concentrations, the upconversion intensity decreased rapidly. The reason of the decrease would be due to remarkable non-radiative relaxation through concentration and defect-related quenching as oxygen vacancies may increase at higher Ni concentrations.41 The energy transfer efficiency is expected to be independent of the Ni2+ concentrations because the Ni2+ concentration is very low compared to the Er3+ concentrations. Thus increased Ni2+ absorption enhanced the overall energy transfer and hence intensified the Er3+ upconversion. However, effects of high absorption and energy transfer efficiency were overcome by nonradiative loss at a relatively higher Ni2+ concentration as mentioned above and reduced the overall upconversion intensity. Inset in the Fig. 5 shows the effect of Nb5+ concentration on the upconverted emission integrated over 950–1050 nm. By introducing proper amount of Nb5+ ions (twice the amount of Ni2+) the upconverted emission intensity was almost doubled. Since, Ni2+ ions were introduced at the Zr4+ sites, charge imbalance reduced the solubility of Ni2+ ions into the CaZrO3 host (no Ni-phase was detected due to low doping concentration) or stabilised the Ni3+ ions to reduce the stress of charge imbalance. Substitution of two Nb5+ ions at Zr4+ sites for each Ni2+ addition balanced the total charge that might have increased the Ni-solubility as well as stabilize the Ni2+. As a result, improved upconverted emission intensity was detected.
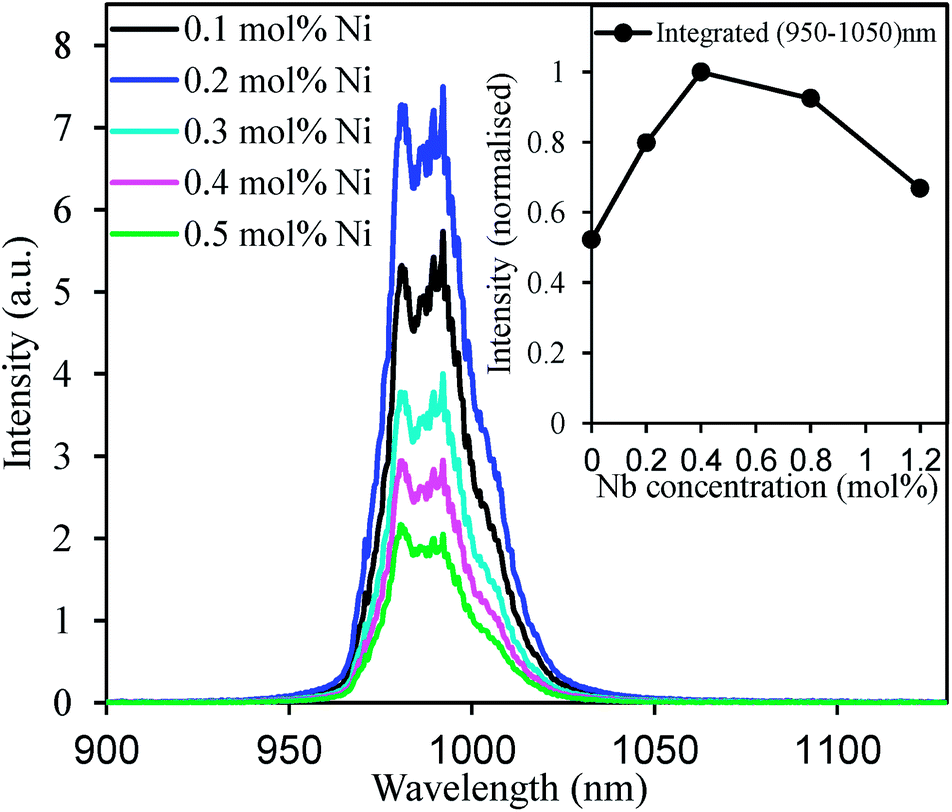 | ||
| Fig. 5 Upconversion spectra of the CaZrO3:15 mol% Er codoped with different mol% of Ni excited at 1300 nm. Inset is the effect of Nb5+ doping concentrations on the upconverted emission intensity (integrated over 950–1050 nm) of CaZrO3:15 mol% Er3+, 0.2 mol% Ni2+ samples. | ||
To evaluate the Ni → Er energy transfer rate quantitatively, time-resolved decay profiles were recorded. Typically, the Ni2+ emission at 1400 nm was recorded for the Ni only doped (no Er) and Ni, Er-codoped (0.2 mol% Ni, 10 mol% Er) samples under the 1180 nm pulsed excitation. The results are presented in Fig. 6. The time-dependent Ni2+ emission intensity of the Ni only doped (no Er) sample INi(no Er)(t) was well fitted using a double exponential eqn (1) mentioned below:39
INi(no Er)(t) = aNi![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp[−t/τ(1)Ni] + (1 − aNi)exp[−t/τ(2)Ni] exp[−t/τ(1)Ni] + (1 − aNi)exp[−t/τ(2)Ni] | (1) |
 | ||
| Fig. 6 Decay profiles of the Ni2+ emission at 1400 nm for the Ni (Nb) only doped (no Er) and Ni, Er (Nb, Li) codoped CaZrO3 samples under the 1180 nm pulsed excitation. Red dotted lines are the results of fitting. | ||
The Ni2+ emission lifetimes τNi are determined by
| τNi = (aNiτ(1)Ni2 + (1 − aNi)τ(2)Ni2)/(aNiτ(1)Ni + (1 − aNi)τ(2)Ni) | (2) |
From the fitting data, τNi was found to be ∼0.6 ms for 0.2 mol% Ni (Nb) only doped CaZrO3, which is comparable with the previously reported values for other oxide hosts.47,48
When the Er3+ acceptors were introduced, the Ni2+ emission intensity decreased more rapidly, because of the energy transfer from the Ni2+ donors to the Er3+ acceptors. The effect of the energy transfer on the time evolution of the emission intensity of the Ni2+ donors is expressed as  when energy migration among the donors is negligibly weak,38,39 whereas it is described by an exponential function exp[−wNi→Ert] for significant migration.49–51 In the present case, the acquired data are fitted by the former better than the latter, i.e.,
when energy migration among the donors is negligibly weak,38,39 whereas it is described by an exponential function exp[−wNi→Ert] for significant migration.49–51 In the present case, the acquired data are fitted by the former better than the latter, i.e.,
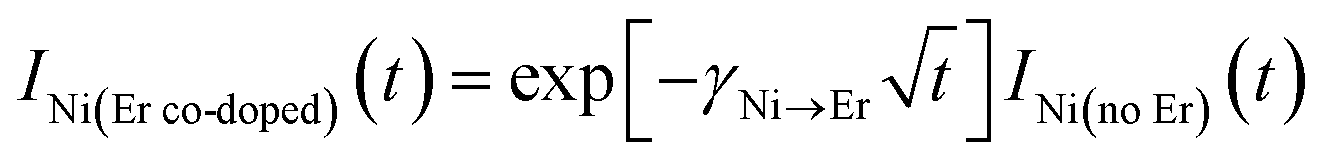 | (3) |
This is consistent with the facts that the Ni concentration is as low as 0.2 mol% and the energy transfer is completed within sub-milliseconds. Using eqn (1) and (3), the energy transfer efficiency ηNi→Er is derived as follows,
 | (4) |
The efficiency obtained from the time-resolved data was about 86%, which is very close to the result obtained from the steady-state Ni-emission intensities as mentioned earlier.
Fig. 7 shows the effect of monovalent ions (Li+, Na+, K+) substitution on the upconversion intensity of the Er, Ni codoped CaZrO3 upconverters. Addition of equivalent amount of alkali ions to that of doped Er3+ ions remarkably increased the upconversion intensity. Because the Er3+ ions (0.106 nm) were substituted for the Ca2+ (0.112 nm) sites, there was imbalance of the total charge. As a result, stress was produced that limits the solubility of the Er3+ ions in CaZrO3 host, which was confirmed by the existence of segregated Er2O3 impurity phase in the XRD patterns as stated previously (Fig. S2†). When the equivalent amount of monovalent ions such as Li+, Na+, K+etc. were added, the charge neutrality was maintained, and consequently the Er3+ impurity phase was remarkably reduced (see Fig. S2†). There were no significant differences in the absorption peak positions of the Er3+ and Ni2+ but remarkable enhancement of the peak intensities around the Er3+ and Ni2+ absorption bands was observed (see Fig. S6†). The enhanced absorption of Er3+ is obviously due to increased numbers of the Er3+ ions inside the CaZrO3 lattice, resulting in more intense upconversion. However, the upconversion intensity was found in the order of Li+ ≫ K+ > Na+ > no alkali ions, and could not be explained by charge compensation only. The f–f electronic transitions of rare earth ions are partially forbidden. As a result, they exhibit low transition probabilities in presence of inversion symmetry.44 It has been reported that substitution of larger or smaller ions into host cation sites remarkably distorts the crystal geometry from the ideal symmetry and hence changes the crystal field splitting.44 As a result, the f–f transition probabilities increase and absorption originating from the Er3+ pronounce. The similar trend was observed in the present case too, where the Er3+ absorption bands were pronounced more by the substitution of the smaller Li+ ions (0.092 nm) at the Ca2+ sites (0.112 nm) than the similar sized Na+ ions (0.118 nm). Substitution with K+ ions (0.151 nm) is also expected to intensify the upconversion as with Li+ ions, however, K+ ions are much larger than Ca2+ ions and it is difficult to occupy the Ca2+ sites. Thus, some of the added K+ ions (hence the same number of the Er3+ ions) did not exist inside the CaZrO3 crystals and remained at the surfaces. Nonradiative transitions from surface ions (here Er3+ ions) will be pronounced due to rapid migration of excited energy to the surface defects.52 Thus, lower Er3+ concentration inside the CaZrO3 lattice and existence of considerable amount of Er3+ ions at the surfaces made the K+-substituted samples less efficient compared to the Li+-substituted samples. Furthermore, the K+-substituted sample exhibited lower crystallinity as seen by weaker XRD peak intensity depicted in Fig. S2.† It is well known that luminescence is crystallinity dependent; higher the crystallinity the better is the luminescence performance.
 | ||
| Fig. 7 Upconversion spectra of CaZrO3:Er3+,Ni2+ codoped with alkali ions excited at 1180 nm. Inset is the variation of integrated upconversion intensity over 950–1050 nm with Li+, Na+, K+ ions substitutions. | ||
Fig. 8 shows the absorption, excitation and upconverted emission spectra of the CaZrO3:15 mol% Er(Li), 0.2 mol% Ni(Nb) sample. The excitation spectrum was obtained from the upconverted emission intensity normalized by the square of the excitation photon flux (due to two-photon process), namely, the emission intensity at a constant excitation photon flux. The upconverter developed here provides broadband sensitivity ranging from 1060 nm to longer than 1600 nm as seen from the excitation spectrum. It further suggests that quite strong upconversion was achieved at the Er-absorption band while comparatively weak at the Ni-absorption band, which is in accordance to the absorbance at the corresponding wavelengths. We have compared the upconversion intensities of such a broadband sensitive upconverter developed here with the previously reported similar upconverter (see ESI S7†) and concluded that the upconversion emission of the CaZrO3:Er,Ni-based upconverter is about 1.7 times stronger. Further, the new upconverter developed here can efficiently absorb 1350–1450 nm light even though the solar irradiation at these wavelengths is weak.
 | ||
| Fig. 8 Illustration of broadband-sensitive upconversion in the CaZrO3:Ni2+,Er3+ upconverter along with the Ni and Er stokes emissions excited at 1180 nm. | ||
The photon flux in the solar spectrum integrated over the Er3+ absorption band (1450–1600 nm) is about 2.0 × 1020 m−2 s−1, which is equivalent to ∼1.93 mA cm−2 current density gain for c-Si solar cells assuming perfect upconversion.45 Introduction of the Ni2+ sensitizers increases the absorbed photon flux by 6.0 × 1020 m−2 s−1 (1060–1450 nm), leading to an additional gain of ∼5.94 mA cm−2 that is more than 3 times the gain originating from the Er3+ absorption alone. The optimum current density gain of present silicon solar cell is about 40 mA cm−2.53 Assuming perfect absorption and upconversion, the newly developed CaZrO3:Ni2+,Er3+ upconverter can contribute ∼7.3 mA cm−2 current density gain of present c-Si solar cells that corresponds to nearly ∼18% increase in gain. However, to realize such efficient broadband-sensitive upconverters, and hence higher c-Si solar cell efficiency, further improved Ni2+ and Er3+ absorption is essential and it can be achieved, for example, by coupling with plasmons at the desired wavelengths and such experiments will be conducted in near future.
5. Conclusions
To improve conversion efficiency of c-Si solar cells, we have developed a new class of broadband-sensitive upconverter, CaZrO3:Ni2+,Er3+ that absorbs photons in a broad wavelength range viz. 1060–1600 nm and emits photons at 980 nm to be efficiently absorbed by c-Si solar cells. We used 6-coodinated Ni2+ located at the octahedron centers (Zr4+-sites) and 12-coordinated Er3+ located at the Ca2+ sites, such that efficient energy transfer from the Ni2+ to the Er3+ was achieved. Further, optimization of the Ni2+ and Er3+ concentrations and addition of charge compensators to reduce the stress caused by the Er3+ and Ni2+ doping significantly improved the upconversion efficiency. Assuming ideal upconversion, the newly developed CaZrO3:Ni2+,Er3+ upconverter can contribute ∼7.3 mA cm−2 current density gain of present c-Si solar cells (∼40 mA cm−2) which corresponds to nearly ∼18% increase in gain. Such broadband-sensitive absorption of sub-bandgap photons above the absorption edge of c-Si solar cells and upconverting them to the most sensitive range (980 nm) of the cell offer a clear route towards surpassing the limiting conversion efficiency of single-junction solar cells, and further enable new applications in sub-bandgap detector sensitization and many more.Acknowledgements
This work was partially supported by Advanced Low Carbon Technology Research and Development Program (ALCA), Japan Science and Technology Agency.References
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- T. Markvart, Phys. Status Solidi A, 2008, 205, 2752–2756 CrossRef CAS.
- V. Badescu and A. M. Badescu, Renewable Energy, 2009, 34, 1538–1544 CrossRef CAS.
- J. Wild, A. Meijerink, J. K. Rath, W. G. J. H. M. Sark and R. E. I. Schropp, Energy Environ. Sci., 2011, 4, 4835 Search PubMed.
- X. Huang, S. Han, W. Huang and X. Liu, Chem. Soc. Rev., 2013, 42, 173–201 RSC.
- H. N. Luitel, R. Chand and T. Watari, Displays, 2016, 42, 1–8 CrossRef CAS.
- H. N. Luitel, K. Ikeue, R. Okuda, R. Chand, Y. Mitsunori, T. Torikai and T. Watari, Opt. Mater., 2014, 36, 591–595 CrossRef CAS.
- J. C. Goldschmidt and S. Fischer, Adv. Opt. Mater., 2015, 3, 510–535 CrossRef CAS.
- A. Shalav, B. S. Richards, T. Trupke, K. W. Krämer and H. U. Güdel, Appl. Phys. Lett., 2005, 86, 10–13 CrossRef.
- S. Fischer, R. Martín-Rodríguez, B. Fröhlich, K. W. Krämer, A. Meijerink and J. C. Goldschmidt, J. Lumin., 2014, 153, 281–287 CrossRef.
- G. E. Arnaoutakis, J. Marques-Hueso, A. Ivaturi, S. Fischer, J. C. Goldschmidt, K. W. Krämer and B. S. Richards, Sol. Energy Mater. Sol. Cells, 2015, 140, 217–223 CrossRef CAS.
- J. L. Wu, F. C. Chen, S. H. Chang, K. S. Tan and H. Y. Tuan, Org. Electron., 2012, 13, 2104–2108 CrossRef CAS.
- G. B. Shan and G. P. Demopoulos, Adv. Mater., 2010, 22, 4373–4377 CrossRef CAS PubMed.
- Y. Chen, J. Zhou, Y. Jiao, W. He, H. Wang, X. Hao, J. Lu and S. Yang, J. Lumin., 2013, 134, 504–507 CrossRef CAS.
- J. de Wild, T. F. Duindam, J. K. Rath, A. Meijerink, W. G. J. H. M. van Sark and R. E. I. Schropp, IEEE Journal of Photovoltaics, 2013, 3, 17–21 CrossRef.
- N. C. Dyck and G. P. Demopoulos, RSC Adv., 2014, 4, 52694–52701 RSC.
- J. A. Briggs, A. C. Atre and J. A. Dionne, J. Appl. Phys., 2013, 113, 124509 CrossRef.
- C. M. Johnson, S. Woo and G. J. Conibeer, IEEE Journal of Photovoltaics, 2014, 4, 799–806 CrossRef.
- W. Zou, C. Visser, J. A. Maduro, M. S. Pshenichnikov and J. C. Hummelen, Nat. Photonics, 2012, 6, 560–564 CrossRef CAS.
- S. Ye, E. H. Song, E. Ma, S. J. Zhang, J. Wang, X. Y. Chen, Q. Y. Zhang and J. R. Qiu, Opt. Mater. Express, 2014, 4, 638 CrossRef.
- A. Nattestad, Y. Cheng, R. Macqueen, T. Schulze, F. Thompson, A. Mozer, B. Fu, T. Khoury, M. Crossley, K. Lips, G. Wallace and T. Schmidt, J. Phys. Chem. Lett., 2013, 4, 2073–2078 CrossRef CAS PubMed.
- T. F. Schulze and T. W. Schmidt, Energy Environ. Sci., 2014, 8, 103–125 Search PubMed.
- T. F. Schulze and T. W. Schmidt, Energy & Environmental Science, 2015, 8, 103–125 CAS.
- C. Strümpel, M. McCann, C. Del Canizo, I. Tobias and P. Fath, Proc. 20th European Photovoltaic Solar Energy Conference, Barcelona, Spain, 2005, pp. 43–43 Search PubMed.
- T. Suzuki, G. S. Murugan and Y. Ohishi, J. Lumin., 2005, 113, 265–270 CrossRef CAS.
- N. Vasileva, P. A. Gerus, V. Sokolov and V. G. Plotnichenko, J. Phys. D: Appl. Phys., 2012, 45, 485301 CrossRef.
- M. Alfredsson, F. Corà, D. P. Dobson, J. Davy, J. P. Brodholt, S. C. Parker and G. D. Price, Surf. Sci., 2007, 601, 4793–4800 CrossRef CAS.
- E. P. Dubrovina, V. A. Sandulenko, M. I. Demchuk, N. V. Kuleshov and V. P. Mikhailov, Chem. Phys. Lett., 1990, 170, 473–477 CrossRef CAS.
- M. Saiful Islam and R. Andrew Davies, J. Mater. Chem., 2004, 14, 86–93 RSC.
- J. Koetke, G. Huber and K. Petermann, J. Lumin., 1991, 48–49, 564–568 CrossRef CAS.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- R. Zhang, H. Lin, D. Chen, Y. Yu and Y. Wang, J. Alloys Compd., 2013, 552, 398–404 CrossRef CAS.
- J. F. Donegan, F. J. Bergin, T. J. Glynn, G. F. Imbusch and J. P. Remeika, J. Lumin., 1986, 35, 57–63 CrossRef CAS.
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS PubMed.
- T. Y. Eeu, X. G. Pang, T. Q. Leow, I. Zuahiri and R. Hussin, Adv. Mater. Res., 2014, 895, 265–268 CrossRef.
- S. Kuck, Appl. Phys. B: Lasers Opt., 2001, 72, 515–562 CrossRef CAS.
- J. F. Suyver, A. Aebischer, D. Biner, P. Gerner, J. Grimm, S. Heer, K. W. Krämer, C. Reinhard and H. U. Güdel, Opt. Mater., 2005, 27, 1111–1130 CrossRef CAS.
- T. Förster, Ann. Phys., 1948, 437, 55–75 CrossRef.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Third edn, Springer, New York, 2006 Search PubMed.
- R. Martín-Rodríguez, S. Fischer, A. Ivaturi, B. Froehlich, K. W. Krämer, J. C. Goldschmidt, B. S. Richards and A. Meijerink, Chem. Mater., 2013, 25, 1912–1921 CrossRef.
- Y. Takeda, S. Mizuno, H. N. Luitel and T. Tani, Appl. Phys. Lett., 2016, 108, 04390 CrossRef.
- J. Fu, Q. Zhang, Y. Li and H. Wang, J. Alloys Compd., 2009, 485, 418–421 CrossRef CAS.
- R. Grimes, Atomistic Simulation, http://abulafia.mt.ic.ac.uk/shannon/ptable.php Search PubMed.
- Q. Huang, H. Yu, E. Ma, X. Zhang, W. Cao, C. Yang and J. Yu, Inorg. Chem., 2015, 54, 2643–2651 CrossRef CAS PubMed.
- Solar spectrum AM1.5, http://rredc.nrel.gov/solar/spectra/am1.5/.
- Materials Project, https://www.materialsproject.org.
- B. Wu, J. Qiu, E. Wu and H. Zeng, Opt. Mater., 2013, 35, 983 CrossRef CAS.
- G. Dong, M. Liang, H. Qin, G. Chai, X. Zhang, Z. Ma, M. Peng and J. Qiu, Phys. Chem. Chem. Phys., 2012, 14, 13594 RSC.
- J. A. Caird, A. J. Ramponi and P. R. Staver, J. Opt. Soc. Am. B, 1991, 7, 1391–1403 CrossRef.
- A. I. Burshteĭn, Soviet Physics–JETP, 1972, 35, 882 Search PubMed.
- C. Parent, C. Lurin, G. Le Flem and P. Hagenmuller, J. Lumin., 1986, 36, 49–55 CrossRef CAS.
- S. Som, A. K. Kunti, V. V. Kumar, S. Dutta, M. Chowdhury, S. K. Sharma, J. J. Terblans and H. C. Swart, J. Appl. Phys., 2014, 115, 193101 CrossRef.
- M. A. Green, K. Emery, Y. Hishikawa, W. Warta and E. D. Dunlop, Prog. Photovoltaics, 2016, 24, 3–11 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra10713c |
| This journal is © The Royal Society of Chemistry 2016 |