
Effective masses, electronic and optical properties of (111)-layered B-site deficient hexagonal perovskite Ba5M4O15 (M = Ta, Nb): a DFT study using HSE06

Abstract
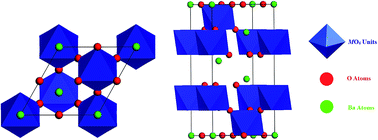
Equilibrium lattice parameters, electronic structures and optical properties of (111)-layered B-site deficient hexagonal perovskite Ba5M4O15 (M = Ta, Nb) were studied by first-principles computations on the basis of density functional theory using the norm-conserving-type pseudo-potential technique and screened nonlocal exchange-correlation functional HSE06 as defined by Heyd, Scuseria, and Ernzerhof. The calculated band dispersions showed that Ba5Ta4O15 and Ba5Nb4O15 are indirect band gap materials (A → G) with band gaps of 3.81 and 3.56 eV, respectively. The effective masses of photogenerated electrons and holes for Ba5Ta4O15 and Ba5Nb4O15 were evaluated in two principal directions at the G (Gamma) point. The Ta–O and Nb–O bonds in the MO6 octahedral environments have polar covalent nature due to the p–d hybridization between O-2p and Ta-5d or Nb-4d orbitals. Since the valence and conduction bands of Ba5Ta4O15 and Ba5Nb4O15 mainly consist of O-2p and Ta-5d or Nb-4d states, changes in the structure of the MO6 octahedral units can be effective for the band gap energy and consequently photocatalytic activity of Ba5Ta4O15 and Ba5Nb4O15. The optical analysis revealed that the main peak of the imaginary part of the complex dielectric function of Ba5Ta4O15 and Ba5Nb4O15 corresponds to the interband electronic transition from O-2p to Ta-5d or Nb-4d. Also, anisotropies in the effective masses of photogenerated charge carriers and static dielectric tensors of Ba5Ta4O15 and Ba5Nb4O15 in an arbitrary crystallographic direction are presented. High photocatalytic activity of Ba5Ta4O15 and Ba5Nb4O15 for hydrogen generation from water splitting and photodegradation of organic pollutants and/or dye molecules under UV light is related to the light effective masses of photogenerated charge carriers. For the efficient solar-energy conversion, the electronic band structures, such as band-edge position and band gap, of Ba5Ta4O15 and Ba5Nb4O15 can be tuned by doping.
 Please wait while we load your content...
Please wait while we load your content...

