
First-principles study of electronic transport and optical properties of penta-graphene, penta-SiC2 and penta-CN2

Abstract
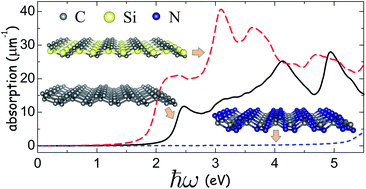
Using density functional theory in combination with the nonequilibrium Green's function formalism we study the electronic transport properties, optical properties and atomic partial charges of the recently proposed isostructural materials: penta-graphene (PG), pentagonal silicon dicarbide (p-SiC2) and pentagonal carbon nitride (p-CN2). Enhanced electronic transport is obtained in p-SiC2 as compared to PG due to the delocalization of the electronic states and smaller variations of the electrostatic potential. This enhancement occurs despite a smaller contribution of Si atoms to the density of states of the system. Penta-SiC2 also displays improved dielectric and optical properties as compared to its all-carbon analogue. For example, larger absorption is obtained in both the visible and the ultraviolet spectral ranges. Strong variation in the atomic partial charge distribution was found in p-SiC2. On the contrary, p-CN2 was not found to exhibit improved optoelectronic properties compared to PG, except for larger partial charges on the surface of the sample. Our findings demonstrate the potential of p-SiC2 in optoelectronic applications.
 Please wait while we load your content...
Please wait while we load your content...

