
Enhanced attachment of human mesenchymal stem cells on nanograined titania surfaces
Jalal Azadmanjiri†
a,
Peng-Yuan Wang†b,
Hitesh Pingleb,
Peter Kingshottb,
James Wanga,
Vijay K. Srivastavac and
Ajay Kapoor*a
aSchool of Engineering, Faculty of Science, Engineering and Technology, Swinburne University of Technology, Hawthorn, Victoria 3122, Australia. E-mail: akapoor@swin.edu.au; Fax: +61 3 9214 8264; Tel: +61 3 9214 8202
bDepartment of Chemistry and Biotechnology, Swinburne University of Technology, Hawthorn, Victoria 3122, Australia
cDepartment of Mechanical Engineering, Indian Institute of Technology, BHU, Varanasi – 221005, India
First published on 26th May 2016
Abstract
Surface nanostructures have shown potential as biomaterials, in tissue engineering and regenerative medicine devices since they have been shown to enhance cellular function by modulating cell–surface interactions in a controlled manner. This work studies human stem cell behavior on titanium dioxide (TiO2) nanotubes that were fabricated on nano-grained (NG) and coarse-grained (CG) substrates. The NG substrates were derived by surface mechanical attrition treatment (SMAT), which has the advantage of being simple to implement. The TiO2 nanotube layer formed on the SMATed titanium (Ti) is thicker and has an inner diameter (70 nm) greater than a comparable layer observed on an untreated (40 nm) substrate. The results illustrate that a NG Ti layer favors the growth of TiO2 nanotubes; presumably due to the high density of grain boundaries and dislocations. An increase in adhesion of human mesenchymal stem cells (hMSCs) in short term culture was observed on the TiO2 nanotubes grown on the NG substrate compared to those grown on the CG substrate, which we attribute to the various roughness and hydrophilicity differences between the two surfaces. Additionally, higher specific strengths of the TiO2 nanotubes may also be achieved by taking advantage of the Ti grain changes on the substrate and the subsequent growth of the nanotubes. Furthermore, structural deformations at the nanoscale can be exploited to manufacture advanced biomaterial surfaces that are designed to enable improved stem cell attachment.
1. Introduction
Surface modification plays a key role in enhancing the biological properties of biomaterials, such as osseointegration, as well as the long-term mechanical stability of Ti or Ti alloy implants.1,2 The topographical length scales of NG and nanostructured materials have been shown to be similar to bone. Therefore, it is hypothesised that NG materials will be promising candidates to improve the biological responses of bone forming cells.3,4 Additionally, other surface properties of a material, such as topography and chemical composition are important in modulating cell–material interactions and the behavior of a diverse range of cells.5–10Despite the breakthroughs made to date in improving the biocompatibility of Ti alloys and TiO2 implants there is still a need for further improvements to such materials that come into contact with biological tissue in hip or dental implants.11 Diverse nanostructured TiO2 morphologies, e.g. nanotubes, rods, wires, etc., have been generated by various fabrication methods, such as hydrothermal, electrochemical fabrication and surfactant templating.11–13 These methods have been shown to produce ideal structures and compositions of TiO2 based materials with novel properties and applications in the fields of chemical sensors, optics, electronics and biomedical implants.14–16
Nanoscale geometry of TiO2 nanotubes with the unique architectures of high surface to volume ratio and controllable dimensions making this material attractive for biomedical applications.17–19 Former studies have fabricated TiO2 nanotubes with different sizes from 15–100 nm diameter.17,18,20,21 For example, Brammer et al.17 studied primary bovine aortic endothelial cells (BAECs) behavior on the untreated Ti surface and TiO2 nanotube with ∼70 nm inner diameter. They have demonstrated that motile cell protrusions are able to probe down into the nanotube pores for contact stimulation, and focal adhesions are formed and disassembled readily for enhanced advancement of cellular fronts. But this was not observed on the untreated Ti substrate. In another study by Park et al.18 TiO2 nanotube with >50 nm diameter reduced the cellular activity and a high extent of programmed cell death of rat bone marrow MSCs. More recently, Lv et al.19 studied adipose-derived stem cells (ADSCs) on nanotubes with different diameter and demonstrated that TiO2 nanotubes with a diameter of 70 nm is the optimal dimension for the osteogenic differentiation of hASCs.
Previous studies also demonstrated that the surface modification of TiO2 nanotubes is favorable at improving their biocompatibility through the introduction of new functionalities, such as generation of a chemically active layer [poly(sodium styrenesulfonate) (PSS) and poly(ethylene glycol) (PEG)],22 SrTiO3 decoration23 and embedding of antibacterial agents (e.g. silver).24 In addition, a NG Ti surface is conducive to the growth of TiO2 nanotubes for fabrication of such surfaces.25 The effect of the Ti substrate structure, such as grain size and crystal defects, on the anodization formation of TiO2 nanotubes has been studied by Zhang and Han.25
Other types of surface nanostructures have been fabricated for stem cell culture including nanoporous structures. For example, porous silicon (pSi) and alumina (pAl) have been fabricated using electrochemical etching for hMSC culture and differentiation.26–28 One key finding from these studies was that both the surface nanoroughness and pore size of these surfaces is crucial for stem cell adhesion. Both the pore size and the properties between pores (nanoroughness and spacing) heavily influenced cell adhesion and subsequent behavior such as osteogenic differentiation.
Lv et al.19 study shows that a surface with 70 nm diameter nanotubes is better than one with 40 nm diameter nanotubes in terms of adhesion and proliferation. Our work utilises SMAT to increase the diameter of nanotubes from 40 to 70 nm. However, there is no published work on TiO2 nanotubes formed on a NG surface and subsequent studies on cellular responses. Therefore, in this study the effect of surface nanocrystalline structure as well as fabricated TiO2 nanotubes on the NG structures has been investigated with regard to adhesion of human adipose-derived stem cells. This report shows that the method we used to fabricate such structures is versatile and simple and is able to enhance the attachment of stem cells, which we attribute to the unique surface properties of the NG TiO2 nanotubes. This current study also lays the foundation concerning the growth of TiO2 nanotubes on a NG structured substrate that includes investigation of the cellular responses. The results examine topology effects on cell responses for the development of new biomaterial surfaces.
2. Materials and methods
2.1. Materials
The as-received commercially pure (CP) Ti (Grade 2, Ningbo Galaxy International Trading Co., Ltd, China) discs of 8 mm diameter and 2 mm thickness were procured as substrates by electro-discharge machining billets. The Ti foil (Nanjing Suntech Metal Products Co, Ltd, China) (99.6% purity) was of thickness 0.25 mm. Ceramic zirconia oxide balls (ZrO2, Grade 5) with 3 mm diameter were purchased from Hong Kong (NanJing Modern Electronic Co., Ltd). Ammonium sulfate [(NH4)2SO4, ≥99.0% purity] and ammonium fluoride (NH4F, ≥99.99% purity) were obtained from Sigma Aldrich (Australia). A platinum counterelectrode with a 1 cm2 surface area in a two-electrode configuration using a direct current (DC) power supply (Powertech MP3086) was employed.2.2. Surface mechanical attrition treatment (SMAT) and substrate preparation
The as-received Ti discs samples were ground to remove the oxide layer using a series of silicon carbide papers, then cloth polished with a colloidal silica suspension to a mirror finish. Subsequently the discs were degreased by sonication in acetone, methanol and ethanol for 15 min each, respectively. The final preparation involved a wash with deionized water and drying in a stream of nitrogen gas. Samples were then treated using a SMAT machine for 0, 20, 40, 60, 80, 100 and 120 min at room temperature (RT). The set-up for the SMAT process has been described in detail elsewhere.29,30 The vibration frequency of the system was 20 kHz and Ti discs were mounted on the top while ZrO2 balls with a diameter of 3 mm were placed at the bottom of a cylinder-shaped chamber. The bottom of the chamber was vibrated at 20 kHz; resulting in the balls resonating and impacting the surface of the Ti discs. After the SMAT process all samples were ultrasonically cleaned in acetone, ethanol and distilled water for 10 min, i.e. a total wash time of 30 minutes, before anodization and characterization of the microstructure and surface properties. The surface of as-received and SMATed samples were etched in 4 ml hydrofluoric acid (HF, 60% concentration) and 96 ml distilled water, at RT, to examine the surface microstructure.2.3. Fabrication of TiO2 nanotubes via anodization
The SMATed and as-received Ti discs were spot-welded to Ti foil to form the anode of an electrochemical cell. The samples were placed 4 cm from the platinum counter electrode with a 1 cm2 surface area in a two-electrode configuration using a DC power supply (Fig. 1). TiO2 nanotubes were fabricated on the surface of SMATed and as-received Ti discs via anodization at room temperature with the electrolyte composed of 1 M (NH4)2SO4 with the addition of 0.5 wt% NH4F. The electrolyte was stirred continuously at 20 rpm using a magnetic stirrer. The applied voltage was fixed at 20 V during the anodization process and the Ti samples were treated for 2 h.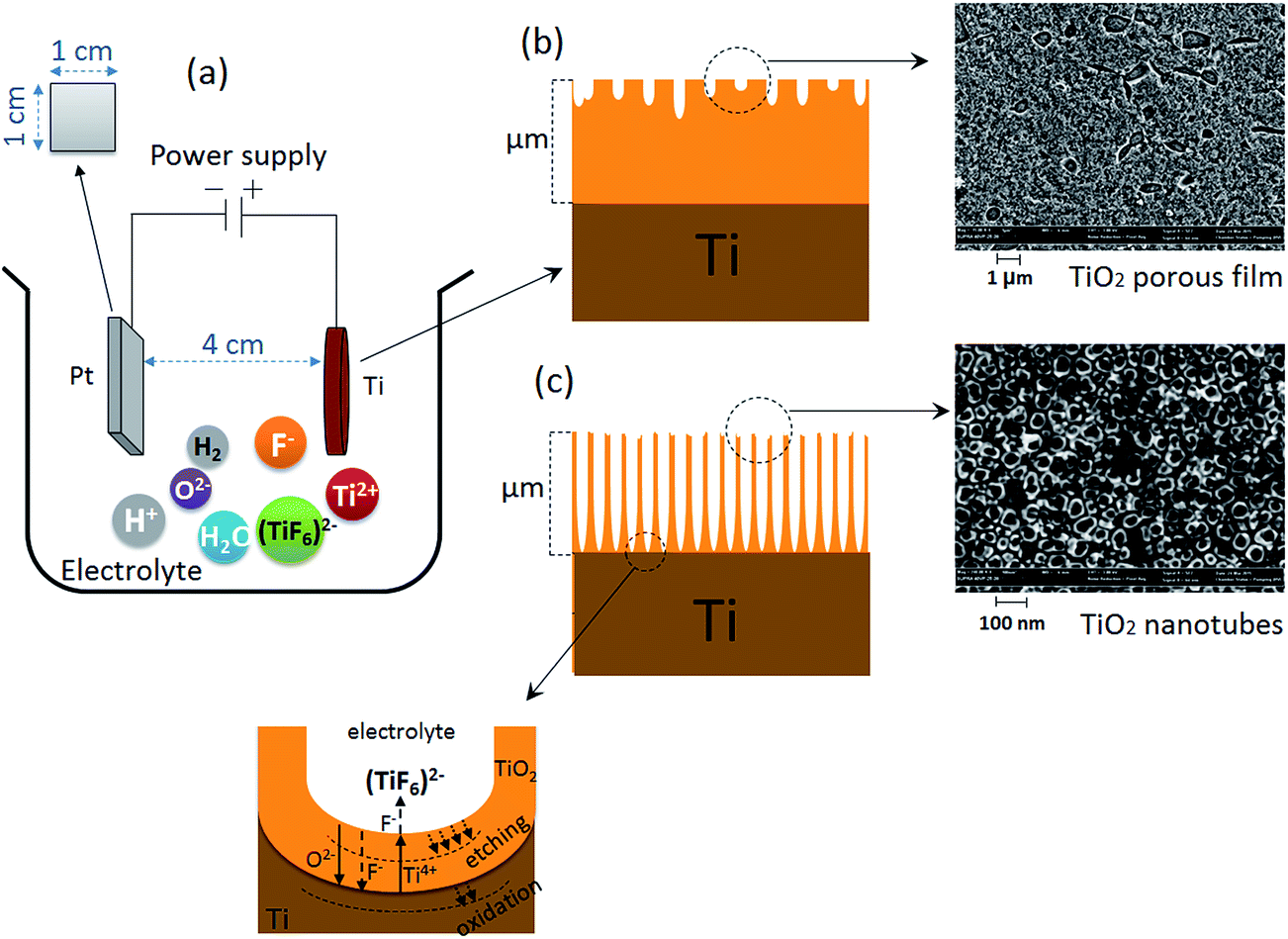 | ||
| Fig. 1 (a) Illustration of an electrochemical cell that indicates the electrolyte ions species, (b) schematic and SEM image of top view of nanoporous TiO2 fabricated on a CP-Ti after anodization for 30 min in 0.5 wt% NH4F, 20 V, and (c) schematic and SEM image of top view of TiO2 nanotubes fabricated on CP-Ti after anodization for 2 h in 0.5 wt% NH4F, 20 V. | ||
After the electrochemical treatment, the obtained samples were immediately washed with Milli-Q water for 5 min and dried at room temperature under a nitrogen gas stream. After anodization, the samples were annealed at 500 °C for 3 h at a heating rate of 2 °C min−1 in a conventional muffle furnace (Nabertherm LT15/13/P330; Nabertherm GmbH, Lilienthal, Germany). The cross section of the samples were mechanically polished using silicon carbide paper to grade 2500, then with a polishing cloth with a colloidal silica suspension, and finally etched in a solution consisting of 10 ml HF (30% concentration), 35 ml HNO3 (70% concentration), and 55 ml distilled water, at RT for 55 seconds.
2.4. Surface characterization
Phase characterization was performed by X-ray diffraction (‘Bruker Advance D8’ from Bruker Pte. Ltd., Singapore) to assess the Ti phases, prior to and after the SMAT and anodization processess. The samples were analyzed with CuKα (λ = 0.154 nm) radiation with 2θ between 20 and 80° and a step size of 0.02°. Results were then compared with the JCPDS powder diffraction standard to confirm the presence of Ti and any the presence of other compounds. The surface element composition was determined using X-ray photoelectron spectroscopy (XPS, AXIS Nova, Kratos Analytical Ltd, Manchester, UK). Samples were cleaned with acetone and ethanol and dried using N2 gas. Survey spectra (0–1200 eV; pass energy 160 eV) were acquired at a power of 10 mA/HT 15 kV = 150 W and were used to identify and determine the percentage of elements on each surface. Two surfaces with 2 points on each surface were analysed (n = 4) and the measurement were repeated. A field-emission scanning electron microscopy (FESEM, Zeiss Supra 40VP) was used to characterize the morphology of the surfaces. At least four random locations on each sample surface were selected for analysis.Vickers microhardness measurements were performed on a micro-Vickers/Knoop testing machine (BUEHLER, Lake Bluff, Illinois USA) under ambient conditions at loads of 5 and 10 gf. Surface roughnesses were measured using a 3D profilometer (Bruker, Contour GT-K1; Bruker Pte. Ltd., Singapore). Three dimensional profiles were drawn and analyzed by the installed SurfVision software (Veeco Instruments Inc.; Plainview, NY-USA). The classical roughness parameters (i.e., Ra and Rq) were calculated from the profiles to measure the absolute roughness of the surface topography.
The surface wettability of each surface was determined using a water contact angle (WCA) goniometer (KSV instruments Ltd, Finland) and the static sessile drop method. A drop of Milli-Q water (0.5 μL) was deposited on substrate surfaces and six spots were analyzed and averaged (n = 6).
2.5. Stem cell culture and analysis
Prior to cell seeding, the substrates were sterilized using 70% ethanol and rinsed three times with sterile PBS (phosphate-buffered saline). Primary human adipose-derived stem cells (hADSCs) were passaged and seeded with a cell density of 5 × 103 cells per cm2 for cell growth according to a previous study.31 Cell morphology, density and spreading area were characterized after 24 h, whilst cell growth was determined after 4 days using an inverted fluorescence microscopy (Eclipse Ti-E, Nikon, Japan). The details of cell–surface interactions was observed using field emission SEM (FE-SEM; ZEISS SUPRA 40 VP, Carl Zeiss, Germany). At each time point, samples were rinsed with PBS twice, fixed with 4% paraformaldehyde, and permeated with 0.2% PBST (PBS with Triton X-100). Cell nuclei and F-actin were stained by DAPI (4′,6-diamidino-2-phenylindole, 100 nM) and phalloidin-TRITC (tetramethylrhodamine, 500 nM), respectively, for 1 h. Cell density and cell spreading area was determined by counting nuclei and quantifying F-actin staining, respectively, from 4× fluorescence images using ImageJ software according to previous studies.32,33 At least six images were analysed at each time point (n = 6) and the standard error mean was used for statistical analysis. Statistical analysis was performed using GraphPad Instat 3.0 (La Jolla, CA, USA). All experiments were at least repeated three times. The statistical analysis between each group was determined with one-way ANOVA and Student–Newman–Keuls multiple comparison tests. p < 0.05 was considered a significant difference. The preparation of materials is presented in Fig. 2. | ||
| Fig. 2 Flow chart of the methods for preparing of materials. | ||
3. Results
3.1. Structure, microhardness and formation of TiO2 nanotubes on as-received Ti and SMATed Ti
Fig. 3 shows SEM and optical microscopy (insets) images of the surfaces and cross-sections for the as-received, anodized, SMATed and anodized-SMATed Ti samples. Schematic representations of the microstructures are also shown. The initial average grain size of as-received the Ti sample was 48 μm. Fig. 3a2 and c2 show the cross-sectional SEM images of as-received and SMATed Ti samples. The Ti samples, after SMAT, display a gradient structure with fine grains from the top surface layer to a grain size of ∼48 μm into the matrix. | ||
| Fig. 3 SEM images and optical microstructures (insets), cross section and schematics of the (a) as-received, (b) anodized, (c) SMATed and (d) anodized-SMATed Ti samples. | ||
It can be seen from Fig. 3c2 and c3, that the total severe plastic deformation layer after SMAT treatment is about 75 μm thick. Fig. 3b and d shows the morphologies of the TiO2 layers on the as-received and SMATed Ti anodized for 2 hours, respectively. The inner diameter size distribution of the TiO2 nanotubes were measured by counting approximately 50 nanotubes at different positions for each of two samples using the ZEISS Smart SEM software. The length of the formed TiO2 nanotube layer was also measured from the cross-sectional images. It was found that the inner diameter and length of the formed TiO2 nanotubes for the SMATed Ti sample is larger than that of the as-received Ti (Fig. 3b2, b3, d2 and d3). The average inner diameters for the as-received and SMATed Ti samples is 40 ± 5 nm and 70 ± 5 nm, respectively. The thickness of the TiO2 layer for the as-received Ti sample was around 10 μm and the corresponding layer thickness for the SMATed Ti sample was 18 μm.
Fig. 4 shows the XRD patterns of as-received, anodized, SMATed and anodized-SMATed Ti samples. The XRD patterns of the as-received and SMATed Ti samples both have the same peaks (Fig. 4a and c) that are assigned to Ti. However, the TiO2 (anatase) phase appears in the XRD patterns of anodized and anodized-SMATed Ti samples (Fig. 4b and d).
 | ||
| Fig. 4 XRD patterns of (a) as-received, (b) anodized, (c) SMATed and (d) anodized-SMATed Ti samples. | ||
XPS analysis was performed on the sample surfaces and the elemental composition data are summarised in Table 1. The as-received and SMATed Ti samples exhibit more trace elemental impurities including Na, Zn, Ca, Al, Si, Fe, and B (6–7% total) compared to the anodized samples (1–4% total). It is reasonable to suggest that the SMAT process may contaminate surfaces due to the use of ceramic balls, whereas anodization reduces this contamination. In general, the surface chemistry changed slightly after the SMAT and anodization processes. Carbon contents increased and the oxygen contents decreased after both treatments (Table 1). The Ti content was observed to decrease after SMAT treatment; probably due to carbon contamination after the SMAT process.
| Condition of Ti sample | Element detection | |||
|---|---|---|---|---|
| C 1s | O 1s | Ti 2p | Traces | |
| As-received | 27.3 ± 1.6 | 49.9 ± 0.7 | 15.9 ± 1.5 | 6.9 ± 0.7 |
| Anodized | 38.6 ± 15.8 | 45.0 ± 7.4 | 12.9 ± 8.1 | 3.5 ± 0.4 |
| SMATed | 35.8 ± 2.7 | 47.2 ± 2.2 | 10.9 ± 4.2 | 6.1 ± 0.0 |
| Anodized-SMATed | 40.0 ± 3.2 | 45.0 ± 2.7 | 13.0 ± 0.9 | 1.2 ± 0.5 |
Fig. 5 displays the microhardness data on the SMATed samples fabricated using different SMAT processing times. The microhardness was measured using at least 20 tests at different locations on the sample. The applied loading force was 5 to 10 gf to prevent the underlying Ti substrate from dominating the mechanical properties of the treated layer.
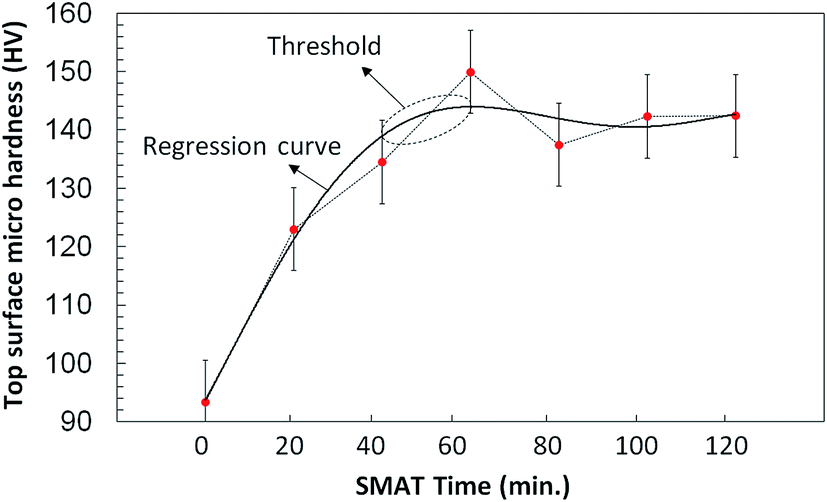 | ||
| Fig. 5 The surface hardness data generated from the microhardness tests, on the as-received and SMATed samples after different SMAT times. Error bar = standard error of mean. | ||
The microhardness for the as-received Ti sample was 93 ± 2 HV and this hardness value increased with increasing SMAT treatment time to reach a maximum of 150 ± 3 HV for the sample with 60 minutes of SMAT processing, after which the microhardness values for the samples with 80, 100 and 120 minutes treatment times remained the same. Fig. 6 shows the Ra roughness parameter for the as-received, anodized, SMATed and anodized-SMATed Ti samples, which were measured using a 3D profilometer. Fig. 6 and inset are indicating of average roughness of the sample surfaces. As it can be observed the average roughness increased when the samples were SMATed and anodized. The anodized-SMATed Ti exhibited an average roughness that is approximately 83% larger than the as-received Ti.
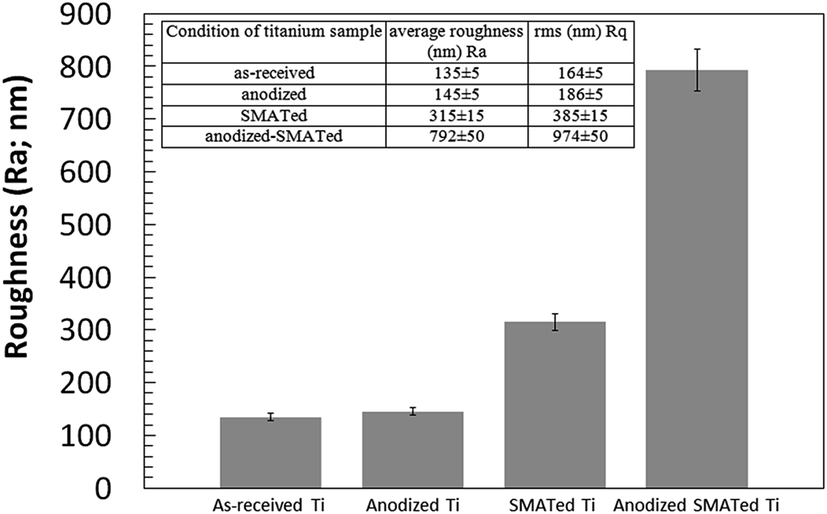 | ||
| Fig. 6 Diagram and inset show roughness parameters (Ra and Rq) for the as-received, anodized, SMATed and anodized-SMATed Ti samples, obtained via 3D profilometer. Error bar = standard error of mean. | ||
The surface wettability of the samples depends on both the surface structure and chemistry (Fig. 7). It was shown that the anodized-SMATed Ti surface had an increase in surface hydrophobicity or decreased wettabilities (WCA ∼ 85) compared to the as-received, anodized and SMATed samples (WCAs ∼ 60).
 | ||
| Fig. 7 Water contact angles of the different surfaces. The sessile drop water contact angle was determined using 5 μL DI water (n = 10). Error bar = standard deviation. | ||
3.2. Stem cell culture
Fig. 8 and 9 show the cell density, cell morphology, averaged cell area and total surface coverage of cells on the surfaces. Cell density on SMATed and anodized SMATed Ti samples were higher than as-received and anodized Ti (Fig. 8). Whereas anodization slightly enhanced cell attachment, SMAT significantly enhanced cell attachment. Cells did not grow fast except on the SMATed Ti sample; which may be due to the late passage of human primary cell used (passage = 6). Cells grew slower and started to differentiate.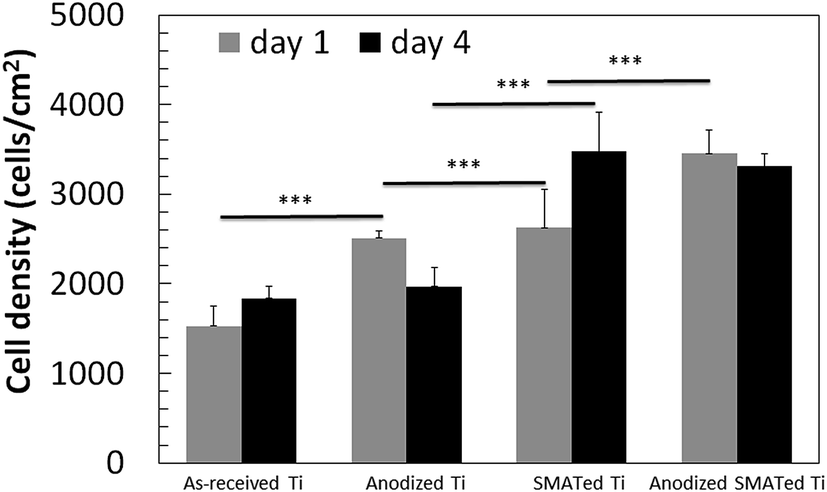 | ||
| Fig. 8 Cell density after 1 and 4 days. Five-ten images of each surface were analysed (n = 5–10). *** indicates p < 0.001. Error bar = standard error of mean. | ||
 | ||
| Fig. 9 (a) Cell morphology after 24 h. F-Actin and nucleus were stained by phalloidin-TRITC and DAPI. Arrows indicate thin and long cell extension. (b) Averaged cell area and (c) total surface coverage of cells after 1 and 4 days. Five-ten images of each surface were analysed (n = 5–10). *, **, and *** indicate p < 0.05, 0.01, and 0.001. Error bar = standard error of mean. | ||
Cells on the as-received Ti spread more than on other surfaces due to the lower roughness; whereas cells on anodized samples (anodized Ti and anodized SMATed Ti) have lower spreading area and exhibit thinner and longer extensions (Fig. 9a). The averaged cell coverage (i.e., coverage/nuclei) revealed the same trend, where cell spreading decreased after SMAT and anodization (Fig. 9b). The total surface coverage on the SMATed Ti sample is, however, the highest, while on the anodized Ti is the lowest (Fig. 9c).
The details of the hADSC interactions with surfaces on all samples was observed using SEM. Fig. 10 shows the spreading of cells on the samples after 4 days of stem cell culture. hADSCs cells attached, spread and grew well on the surfaces of all samples.
 | ||
| Fig. 10 Schematic and SEM images of hADSCs on surfaces (after 4 days) of the (a) as-received, (b) anodized, (c) SMATed and (d) anodized-SMATed Ti samples. Arrows indicate the interaction between filopodia and surface structure. | ||
4. Discussion
Firstly, it is found that the gradient structure with a very fine grain size formed on the top surface layer of SMATed sample is a result of a gradual reduction within the applied strain with increasing depth of the deformed layer. This effect represents the complete range of structural changes throughout the SMAT treatment. It is also observed that the TiO2 nanotubes formed on the SMATed surface tend to have larger dimensions both in diameter and length. In addition, the XRD patterns and XPS analysis of the as-received and SMATed Ti samples both have the same Ti peaks and elements, respectively, thus the SMAT process does not result in any phase transformation of Ti. The dominant elements by XPS characterization are from Ti, O and C in all samples. Presence of oxygen and carbon are due to the quick adsorption of these elements on the surface. As soon as the samples are exposed to the ambient, oxygen and carbon are easily adsorbed on the surface because titanium has a high affinity for oxygen and carbon.34 The larger dimensions in diameter and length of SMATed sample are due to a greater grain boundary and dislocations on the SMATed surface. In fact grain boundaries and dislocations play the principal role in electrolyte ion diffusion and accelerating rate throughout anodization.25 Thus, the very fine grains of the Ti substrate is favorable for the growth of TiO2 nanotubes.It is demonstrated that with increasing SMAT time the hardness value is prolonged and it reaches a maximum value at 60 minutes SMAT treatment and subsequent times do not change the hardness threshold point. The reason for such a different increment of microhardness can be attributed to the refining grain size and higher dislocation density during different SMAT times. Dislocation activities and plastic deformation behaviors in metals and alloys, which depend strongly on the lattice structure and also the stacking fault energy (SFE), lead to increases in the microhardness of samples.29,35 The recent study by Bahl and et al. also showed that generated NG surfaces, processed by the SMAT technique, increase the corrosion fatigue strength of a 316L stainless steel metal alloy, compared to the CG surface.36
Roughness analysis for the as-received, anodized, SMATed and anodized-SMATed Ti samples showed that the anodized-SMATed Ti has the highest roughness. On the other hand, the roughness value difference between the as-received and anodized Ti samples is not great, while there is a large difference between SMATed and anodized-SMATed Ti. This diverse value might be attributed to extensive grain boundaries and dislocations in the anodized-SMATed Ti. The fine grain size and large grain boundaries of Ti are important in assisting with electrolyte ion diffusion and the precipitation rate of anodization. Thus, it can help to increase the roughness values.
WCA represents an overall surface property including topography and chemistry.37,38 There is an optimal WCA for cell adhesion and differentiation, depending on cell types.38 In this study we found that anodized SMATed surface is more hydrophobic than other surfaces. WCA is correlated to both surface chemistry and topography. Cassie–Baxter and Wenzel's models have been developed to describe surface wettability.39 In general, increase of roughness of a hydrophilic surface will decrease the WCA (Wenzel) while increase of roughness of a hydrophobic surface will increase the WCA (Cassie–Baxter). We find that the surfaces in this study behave like Cassie–Baxter model.
In this study, primary hADSCs from lipoaspirate were used. These cell lines are under investigation by the group because of the potential of sourcing adipose tissue from plastic surgery.40 Surface nanotopographies and intrinsic mechanical properties have been shown to influence cell attachment and spreading.18,21,41,42 Cell attachment is the lowest on Ti, but the cells spread more on those samples. This could be due to the space inhibition mechanism. When the cell density is low on the surface, each cell has a larger area to spread. On the other hand, a high cell density will inhibit cell spreading because of cell–cell contacts. On SMATed and anodized SMATed samples, although each cell has less spreading area compared to the flat control possibly due to the increase of roughness, the total cell coverage on SMATed Ti is higher than flat controls. Anodization of as-received and SMATed Ti samples inhibited cell spreading but not cell attachment, indicating that cells are sensitive with the roughness change at the nanoscale. The grown nanotubes on the NG layer may adsorb more proteins, due to higher surface area, and lead to the adsorbed proteins being in a more bioactive conformation compared to the CG surface. Additionally, the reduction of grain size to the nanoscale may accelerate the differentiation of hMSCs but this requires further investigation.
Increases of nanoroughness can be beneficial in some circumstances but not others. However, longer term cellular responses, such as osteogenic differentiation are of interest on these surfaces, and will need to be studied in the future.
5. Conclusion
In this work, a two-step treatment which includes SMAT and anodization has been developed to improve the cell compatibility of Ti. The SMATed surface, after 60 minutes treatment, exhibited the maximum hardness compared to the other SMAT times. This increase in hardness is due to the refining grain size and dislocation density during SMAT time. The average length and diameter of TiO2 nanotubes layer formed on the SMATed Ti are 18 μm and 70 nm, respectively, which are larger than un-SMATed Ti with 10 μm and 40 nm. These larger sizes in SMATed Ti are due to higher acceleration reaction rate and ion diffusion coefficient during anodization, compared to un-SMATed Ti. TiO2 nanotubes on the SMATed Ti also led to an enhancement in stem cell attachment. This increment is attributed to the vastly increase surface roughness on the NG layer and wider nanotubes of the surface. The improved cell attachment and proliferation together with physical and mechanical properties, on the SMAT-processed NG TiO2 nanotubes is a promising surface that could be engaged as a biomaterial for surgical implants.Acknowledgements
The authors acknowledge the financial support through the Australia-India Strategic Research Fund (AISRF) ST060048. The authors are grateful to Prof. C. Wen for providing Ti discs. The Scientific Industrial Endowment Fund (SIEF) and Australia Research Council (ARC) are acknowledged for providing John Stocker Postdoctoral Fellowship (PF12-026) and The Discovery Early Career Researcher Award (DECRA: DE150101755) for PYW. Part of this work was performed at the Biointerface Engineering Hub at Swinburne part of the Victorian Node of the Australian National Fabrication Facility. A company established under the National Collaborative Research Infrastructure Strategy to provide nano and microfabrication facilities for Australia's researchers.References
- S. Tawfick, M. De Volder, D. Copic, S. J. Park, C. R. Oliver, E. S. Polsen, M. J. Roberts and A. J. Hart, Adv. Mater., 2012, 24, 1628–1674 CrossRef CAS PubMed.
- F. Vetrone, F. Variola, P. T. de Oliveira, S. F. Zalzal, J. H. Yi, J. Sam, K. F. Bombonato-Prado, A. Sarkissian, D. F. Perepichka, J. D. Wuest, F. Rosei and A. Nanci, Nano Lett., 2009, 9, 659–665 CrossRef CAS PubMed.
- T. J. Webster, E. L. Hellenmeyer and R. L. Price, Biomaterials, 2005, 26, 953–960 CrossRef CAS PubMed.
- T. J. Webster and J. U. Ejiofor, Biomaterials, 2004, 25, 4731–4739 CrossRef CAS PubMed.
- B. Setzer, M. Bachle, M. C. Metzger and R. J. Kohal, Biomaterials, 2009, 30, 979–990 CrossRef CAS PubMed.
- X. M. Liu, J. Y. Lim, H. J. Donahue, R. Dhurjati, A. M. Mastro and E. A. Vogler, Biomaterials, 2007, 28, 4535–4550 CrossRef CAS PubMed.
- J. Liu, X. D. Wang, Q. M. Jin, T. C. Jin, S. Chang, Z. C. Zhang, A. Czajka-Jakubowska, W. V. Giannobile, J. E. Nor and B. H. Clarkson, Biomaterials, 2012, 33, 5036–5046 CrossRef CAS PubMed.
- Y. Sugita, K. Ishizaki, F. Iwasa, T. Ueno, H. Minamikawa, M. Yamada, T. Suzuki and T. Ogawa, Biomaterials, 2011, 32, 8374–8384 CrossRef CAS PubMed.
- P. Y. Wang, W. T. Li, J. S. Yu and W. B. Tsai, J. Mater. Sci.: Mater. Med., 2012, 23, 3015–3028 CrossRef CAS PubMed.
- P.-Y. Wang, W.-T. Li, J. Yu and W.-B. Tsai, J. Mater. Sci.: Mater. Med., 2012, 23, 3015–3028 CrossRef CAS PubMed.
- G. K. Mor, K. Shankar, M. Paulose, O. K. Varghese and C. A. Grimes, Nano Lett., 2005, 5, 191–195 CrossRef CAS PubMed.
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Langmuir, 1998, 14, 3160–3163 CrossRef CAS.
- D. Gong, C. A. Grimes, O. K. Varghese, W. C. Hu, R. S. Singh, Z. Chen and E. C. Dickey, J. Mater. Res., 2001, 16, 3331–3334 CrossRef CAS.
- K. Shankar, J. I. Basham, N. K. Allam, O. K. Varghese, G. K. Mor, X. J. Feng, M. Paulose, J. A. Seabold, K. S. Choi and C. A. Grimes, J. Phys. Chem. C, 2009, 113, 6327–6359 CAS.
- D. A. Wang, T. C. Hu, L. T. Hu, B. Yu, Y. Q. Xia, F. Zhou and W. M. Liu, Adv. Funct. Mater., 2009, 19, 1930–1938 CrossRef CAS.
- J. Azadmanjiri, C. C. Berndt, J. Wang, A. Kapoor, V. K. Srivastava and C. E. Wen, J. Mater. Chem. A, 2014, 2, 3695–3708 CAS.
- K. S. Brammer, S. H. Oh, J. O. Gallagher and S. H. Jin, Nano Lett., 2008, 8, 786–793 CrossRef CAS PubMed.
- J. Park, S. Bauer, K. von der Mark and P. Schmuki, Nano Lett., 2007, 7, 1686–1691 CrossRef CAS PubMed.
- L. W. Lv, Y. S. Liu, P. Zhang, X. Zhang, J. Z. Liu, T. Chen, P. L. Su, H. Y. Li and Y. S. Zhou, Biomaterials, 2015, 39, 193–205 CrossRef CAS PubMed.
- S. Oh, K. S. Brammer, Y. S. J. Li, D. Teng, A. J. Engler, S. Chien and S. Jin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2130–2135 CrossRef CAS PubMed.
- J. Park, S. Bauer, A. Pittrof, M. S. Killian, P. Schmuki and K. von der Mark, Small, 2012, 8, 98–107 CrossRef CAS PubMed.
- K. Vasilev, Z. Poh, K. Kant, J. Chan, A. Michelmore and D. Losic, Biomaterials, 2010, 31, 532–540 CrossRef CAS PubMed.
- Y. Wang, D. M. Zhang, C. E. Wen and Y. C. Li, ACS Appl. Mater. Interfaces, 2015, 7, 16018–16026 CAS.
- S. L. Mei, H. Y. Wang, W. Wang, L. P. Tong, H. B. Pan, C. S. Ruan, Q. L. Ma, M. Y. Liu, H. L. Yang, L. Zhang, Y. C. Cheng, Y. M. Zhang, L. Z. Zhao and P. K. Chu, Biomaterials, 2014, 35, 4255–4265 CrossRef CAS PubMed.
- L. Zhang and Y. Han, Nanotechnology, 2010, 21, 055602 CrossRef PubMed.
- P. Y. Wang, L. R. Clements, H. Thissen, W. B. Tsai and N. H. Voelcker, Biomater. Sci., 2013, 1, 924–932 RSC.
- P.-Y. Wang, L. R. Clements, H. Thissen, A. Jane, W.-B. Tsai and N. H. Voelcker, Adv. Funct. Mater., 2012, 22, 3414–3423 CrossRef CAS.
- P.-Y. Wang, L. R. Clements, H. Thissen, S.-C. Hung, N.-C. Cheng, W.-B. Tsai and N. H. Voelcker, RSC Adv., 2012, 2, 12857–12865 RSC.
- K. Lu and J. Lu, Mater. Sci. Eng., A, 2004, 375, 38–45 CrossRef.
- K. Lu and J. Lu, J. Mater. Sci. Technol., 1999, 15, 193–197 CAS.
- P. Y. Wang, D. T. Bennetsen, M. Foss, T. Ameringer, H. Thissen and P. Kingshott, ACS Appl. Mater. Interfaces, 2015, 7, 4979–4989 CAS.
- P.-Y. Wang, J. Yu, J.-H. Lin and W.-B. Tsai, Acta Biomater., 2011, 7, 3285–3293 CrossRef CAS PubMed.
- P.-Y. Wang, H.-T. Yu and W.-B. Tsai, Biotechnol. Bioeng., 2010, 106, 285–294 CrossRef CAS PubMed.
- Y. Q. Fu, H. J. Du, S. Zhang and W. M. Huang, Mater. Sci. Eng., A, 2005, 403, 25–31 CrossRef.
- J. Azadmanjiri, C. C. Berndt, A. Kapoor and C. Wen, Crit. Rev. Solid State Mater. Sci., 2015, 40, 164–181 CrossRef CAS.
- S. Bahl, P. Shreyas, M. A. Trishul, S. Suwas and K. Chatterjee, Nanoscale, 2015, 7, 7704–7716 RSC.
- P.-Y. Wang, L. R. Clements, H. Thissen, W.-B. Tsai and N. H. Voelcker, Acta Biomater., 2015, 11, 58–67 CrossRef CAS PubMed.
- H. Ahn, I. Lee, H. Lee and M. Kim, Int. J. Mol. Sci., 2014, 15, 2075 CrossRef PubMed.
- P. Y. Wang, H. T. Yu and W. B. Tsai, Biotechnol. Bioeng., 2010, 106, 285–294 CrossRef CAS PubMed.
- P. Y. Wang, H. Thissen and P. Kingshott, ACS Appl. Mater. Interfaces, 2016, 8, 4477–4488 CAS.
- K. S. Brammer, S. Oh, J. O. Gallagher and S. Jin, Nano Lett., 2008, 8, 786–793 CrossRef CAS PubMed.
- S. Oh, K. S. Brammer, Y. S. Li, D. Teng, A. J. Engler, S. Chien and S. Jin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2130–2135 CrossRef CAS PubMed.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |