
Small molecule therapeutic-loaded liposomes as therapeutic carriers: from development to clinical applications
Jae Yoon Hwangb,
Zibiao Li*a and
Xian Jun Loh*abc
aInstitute of Materials Research and Engineering (IMRE), 3 Research Link, Singapore 117602, Singapore. E-mail: lohxj@imre.a-star.edu.sg; lizb@imre.a-star.edu.sg
bDepartment of Materials Science and Engineering, National University of Singapore, 9 Engineering Drive 1, Singapore 117576, Singapore
cSingapore Eye Research Institute, 11 Third Hospital Avenue, Singapore 168751, Singapore
First published on 20th July 2016
Abstract
While small hydrophilic therapeutic molecules have proved to be exceptionally effective in curing diseases, their in vivo efficacy remains low. Small molecule therapeutics infused in vivo result in uncontrolled bio-distribution that causes dilution and unwanted side effects. Therefore, global research to search for ideal carriers for small molecule therapeutics has gained attention seeking to increase efficiency of targeting desired cells. Liposomes are one class of nanocarriers that could encapsulate both hydrophilic and lipophilic small molecule therapeutics, protect them against physiological degradation and deliver them into targeted cells. In this review, studies done from the past up to the present are summarized, and various methods and mechanisms for encapsulation of small therapeutic molecules in liposomes for targeted delivery and triggered release, as well as their potential in the clinical uses, will be discussed.
1. Introduction
Effective encapsulation of small therapeutic molecules and their delivery is one of the key challenges in the study of pharmaceutics. While small therapeutic molecules are only effective when they successfully penetrate into cells, failure in achieving good encapsulation of these small therapeutic molecules would lead to degradation of the molecules before cellular endocytosis.1 Furthermore, poor delivery will lead to random biodistribution of these small molecules, leading to toxicity where they are not needed, or simply failing to express their effects due to low concentrations.2 Therefore, drug carriers that could protect and direct these drug molecules to targeted areas are highly desired for the enhanced efficacy of small molecule therapeutics.2. Liposome
Currently, a number of techniques have been developed to tackle these limitations; pathogenic carriers, liposomes, micelles and other polymer-based synthetic carriers are some of the examples.3–8 Amongst these categories, liposome is considered the safest can act as non-invasive method of delivery of therapeutics.2,9 A typical liposome contains bilayer of long-chained lipid molecule with one end being polar, such as phospholipids.10 With polar head of the molecules facing the aqueous phase, the bilayer array of molecules allows to form a globule that is stable in aqueous phase and also be able to carry an aqueous payload within. Fig. 1 illustrates the typical liposome structure.11 | ||
| Fig. 1 Structure of liposome.11 | ||
As liposome consists of both lipid and polar portions, it is possible to carry both hydrophilic and lipophilic small therapeutic molecules. It is also advantageous that the liposome's surface has the potential to be engineered to configure its general properties such as stability, encapsulation efficiency, delivery accuracy as well as release efficiency. Liposomes may also be engineered to mimic cellular membrane, increasing chance of capsular fusion without any invasiveness.12 For cancer therapies, nanocarrier molecules like liposome exhibits advantageous properties such as enhanced permeability and retention (EPR) which is effective for delivering the small molecule therapeutics to tumours.13 Furthermore, liposomes has the potential to be engineered to tailor its properties, such as PEGylation, which enables the nanocarrier to be in a “stealth” mode, avoiding being engulfed by phagocytes of the in vivo immune system.14–16 Additionally, modification of liposome have successfully enhanced encapsulation, targeted delivery and release abilities.2
3. Encapsulation and delivery
Encapsulation of hydrophilic small molecule therapeutics could prevent its degradation during storage and transportation.17–19 Small molecule therapeutics may also be absorbed by any part of the body, which could potentially lead to undesirable results.1,2 While hydrophilic small molecule therapeutics are known to have high dissolution within polar solvent during thin layer hydration method to synthesize liposome, the efficiency of encapsulation of small molecule therapeutics itself is low due to larger volume ratio of hydration medium to encapsulated aqueous phase of liposome, which is a limitation during clinical use.10 Previously, Xu et al. established a calculated relation to provide an approximation of encapsulation in the unilamellar liposomes based on the observation of ionic strength being inversely proportional to encapsulation efficiency. Additionally, some preparation steps for liposomal encapsulation for polar small molecule therapeutics were introduced earlier, such as reverse phase evaporation and freeze–thaw cycling methods.20,21 Mildly basic small molecule therapeutics such as doxorubicin could also be encapsulated by liposome with high efficiency via remote loading methods.22,23To achieve acceptable expression of desired effects by the small molecule therapeutics, liposome-encapsulated small molecule therapeutics must reach the target cells at sufficiently high concentration. Therefore, it is ideal that the small molecule therapeutics are encapsulated with maximum concentration with minimal leakage during storage and delivery and effective release at the desired region. However, as the ability of liposome to effectively release the encapsulated small molecule therapeutics is in apparent contradiction to the ability to prevent leakage, careful optimization between two abilities is needed for specific applications.24 For example, to increase storage capacity, high-transition temperature lipids or cholesterol (Chol) were added to liposome formulation, but to achieve effective release, other alternatives have to be explored. Addition of ligands such as antibodies to the surface of the liposome that could respond to surrounding media could be used to increase delivery efficiency and trigger release.25
This paper provides a comprehensive review on each one of these major findings and the most successful strategies for liposome as nanocarriers for small molecule therapeutics. As most of the small molecule therapeutics are usually polar and hydrophilic, it is important to identify specific group of small molecule therapeutics to be included in the review. Based on United States Pharmacopeia (USP) hydrophilic small molecule therapeutics classification, small molecule therapeutics noted as ‘very soluble’ (solubility greater than 1000 mg mL−1) to ‘soluble’ (10 to 33 mg mL−1) was included in this review as hydrophilic small molecule therapeutics. This paper aims to review the most relevant and recent literatures on techniques for formulation of hydrophilic small molecule therapeutic-encapsulating liposomes and discusses their encapsulating, triggered delivery and release efficiencies.
4. Discussion
4.1. Methodology to increase passive encapsulation efficiency
While the simplest form of liposomes have low encapsulation efficiency due to the lower concentration of hydrophilic region compared to the surrounding aqueous phase, modification of liposomal composition and surface functionalization could passively increase the encapsulation efficiency. Likewise, similar effects are also observed when liposomes are synthesized using different methods.26While it is generally understood that the inclusion of Chol to egg PC increases liposome's stability and therefore increase encapsulation efficiency, it may not be applicable for some types of small molecule therapeutics. For instance, cisplatin encapsulated in Chol-containing liposome is so stable that it would not be released from liposome at the cytoplasm of the cell.31 Therefore, for such small molecule therapeutics, Chol-free formulation have to be developed.32 For example, Chol-free liposome was used for encapsulate hydrophilic carboplatin. Both DPPC/N-[carbonyl-2,3-bis(methoxypolyethyleneglycol 1000)]-1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE-PEG1000) and 1,2-distearoyl-sn-glycero-3-phosphorylcholine (DSPC)/DSPE-PEG1000 formulations showed over-time leakage during phase-stability tests. However, the leakages were prevented when these formulations were freeze-dried with sucrose and terhalose, which led to reduction in transition temperature from gel to liquid crystalline phase of the lipid bilayer. This phenomenon is largely due to the increased number of hydrogen bonding between polar head of phospholipid and disaccharides.25,33,34
In the study done by Anumita, et al., different encapsulation efficiency of hydrophilic small molecule therapeutic such as carboplatin was dependent on the thickness of the lipid phase, which is determined by the carbon chain length of the phospholipid. From Anumita's research, higher carboplatin encapsulation was achieved for thinner lipid phase, when compared between DPPC (C16 carbon tails) to DPPC (C18 carbon tails).32 Likewise, Gűrsoy et al. have done experiments using egg PC (C16 and C18 carbon tails), egg PC with Chol and DPPC with Chol liposomes to encapsulate isoniazid and rifampicin which are hydrophilic and lipophilic small molecule therapeutics respectively. The results showed that DPPC with Chol liposomes exhibited the highest encapsulation efficiency for both types of molecules.35
However, some phospholipids such as DSPC is still preferred over shorter carbon-chain phospholipid such as egg PC, DMPC (C14 carbon tails) and DPPC for encapsulation of alendronate due to higher vesicle stability generated from negatively charged surface. Furthermore, DSPC's negative charges repels one another making DSPC ideally free from aggregation due to electrostatic repulsion. This phenomenon will in turn lead to longer half-life duration in vivo compared to other lipid based liposomes.36 This is also somehow beneficial as longer chain leads to higher lipid stability due to greater van der waal's interaction, leading to reduction in small molecule therapeutic leakage.37
Studies done by Schneider et al. claimed that lipid concentration is also a factor that affects the encapsulation of polar small molecule therapeutics in particular to itopride and gadolinium diethylenetriaminepentaacetic acid (Gd-DTPA). In the experiment, Soy-phosphatidycholine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cholesterol:Soy-phosphatidyglycerol (SPC:Chol:SPG) liposome at the molar ratio of 6:3:1 was used to encapsulate itopride and Gd-DTPA. The result for itopride encapsulation was 1.2, 40.2 and 45.1% respectively to liposome concentration. Gd-DTPA encapsulation was done at higher liposome concentration of 100 and 250 mg g−1, and produced efficiency of 37.9 and 63.2% respectively.38 In this test, Schneider also identified that increasing small molecule therapeutic concentration led to opposing effect whereby small molecule therapeutic encapsulation efficiency decreased contrary to expectations.38
:cholesterol:Soy-phosphatidyglycerol (SPC:Chol:SPG) liposome at the molar ratio of 6:3:1 was used to encapsulate itopride and Gd-DTPA. The result for itopride encapsulation was 1.2, 40.2 and 45.1% respectively to liposome concentration. Gd-DTPA encapsulation was done at higher liposome concentration of 100 and 250 mg g−1, and produced efficiency of 37.9 and 63.2% respectively.38 In this test, Schneider also identified that increasing small molecule therapeutic concentration led to opposing effect whereby small molecule therapeutic encapsulation efficiency decreased contrary to expectations.38
Xu et al. produced similar study using antiviral small molecule therapeutic, tenofovir. At low concentration of the phospholipids (<100 mM), increasing lipid concentration led to corresponding increase in encapsulation efficiency due to larger small molecule therapeutic encapsulation capacity driven from increased liposome count. However, higher phospholipid concentrations (>160 mM) did not result in correlating increase in encapsulation efficiency due to increased viscosity, which inhibits the inclusion of the small molecule therapeutics during vesicle-formation.
Compositional modification via addition of polymeric molecules into liposome bilayer is also well-known strategy to increased encapsulation efficiency. Belletti et al. studied a small therapeutic molecule for HIV treatment by using cidofovir-loaded hybrid liposomes that was integrated with polymeric particles (Pps).225 The results showed that the hybrid liposome encapsulating cidofovir expressed higher colloid stability as well as better encapsulating efficiency than conventional liposomes. Through various trials and optimization, the Pps-hybrid liposome increased cidofovir encapsulation from 2.5 to 12% when polylactic acid PLS was used. Following this study, lidocaineoctadecyl-quaternized lysine modified with chitosan and Chol was incorporated to liposome respectively, and the encapsulation efficiency increased by 80 and 70% respectively when compared to non-modified typical liposome. Furthermore, for both modification, slower leakage and release was noted.39
In order to verify the effects of charged liposomes for encapsulation of polar small therapeutic molecules, characterizations using differential scanning calorimetry (DSC) and phosphorous nuclear magnetic resonance (31P-NMR) were used to determine the interactions between small therapeutic molecules and the charged liposome that encapsulates the drug. During the characterization, sumatriptan succinate encapsulated in PC:Chol, diacetyl phosphate (DCP) and SA liposomes were used. Sumatriptan is a positively charged small molecule therapeutic, DCP liposome is negatively charged while SA liposome exhibits positive charges. As shown in Fig. 2, the thermal analysis using DSC revealed that sumatriptan was successfully encapsulated by all three types of liposomes. In SA liposome however, a split peak for sumatriptan indicates that there were additional interaction with SA compared to the rest. This interaction may be directly linked to the liposome-small molecule therapeutic stability, whereby SA in the liposome is able to stabilize the encapsulated small molecule therapeutic better than conventional PC:Chol liposome. SA increased the encapsulation efficiency for sumatriptan compared to PC:Chol liposome.40 However, contrary to the hypothesis that electrostatic interactions would increase stability of small molecule therapeutics and charged liposome complexes leading to increased encapsulation efficiency. Negatively charged DCP liposome, despite showing electrostatic interaction with sumatriptan as shown from DSC peaks, showed lower encapsulation efficiency than positively charged SA liposome. This result therefore indicates that the encapsulation efficiency may not be directly affected by the electrostatic interaction between positively charged small molecule therapeutics and negatively charged DCP liposomes.40
 | ||
| Fig. 2 Thermal analysis for (A) cholesterol–PC (CH), sumatriptan (SUMAT) and CH/SUMAT and (B) diacetyl phosphate (DCP), stearylamine (SA), SUMAT, DCP/SUMAT, SA/SUMAT.40 | ||
 | ||
| Fig. 3 Illustrative summary of hydrophilic small molecule therapeutics encapsulation methods: (A) FT, (B) DRV and (C) REV.50 | ||
Freeze–thaw cycling method could have produced enhanced encapsulation efficiency because multiple cycles of freezing and melting of liposome results in formation of ice crystals between closely packed lamellae of the liposome, physically enlarging the internal volume and therefore increasing the aqueous proportion to lipid of the liposome. This phenomenon causes polar small molecule therapeutics to be more available at the aqueous phase within the liposome leading to higher encapsulation.47 In other studies; for instance, studies done by Chapman et al., further researched on encapsulation using FT method, and revealed that freezing/melting rate, number of FT cycles, solute/liposomal constituent and concentration, and other several factors affects the encapsulation efficiency.48 Another study done by Anzai et al. showed that there was at least 10 to 50 times increased encapsulation volume when FT method was used to prepare liposomes. During the preparation, Anzai identified that small liposomes gradually combined to larger liposome during FT progress, making internal volume greater and therefore increasing encapsulation efficiency. From this study, it was experimentally shown that optimum encapsulation efficiency using FT was obtained when the mixture was frozen with liquid nitrogen or solid CO2 and melted under cool air.49
In 2005, Ueno and Sriwongsitanont experimented liposome preparation by varying the number of cycles of FT, to determine the change in average size of the liposome.51 The results showed that number of FT cycles were inversely proportional to the diameter of the liposome, indicating that encapsulation efficiency decreased with increasing number of FT cycles. On the other hand, FT process resulted in disruption and rearrangement of MLVs to small unilamellar vesicles (SUVs), which exhibited increased liposome stability and therefore increased encapsulation efficiency. The disruption and rearrangement of lamellae created new liposomes which increased chance of encapsulating more small molecule therapeutic molecules.52,53 Therefore, FT cycles have to be carefully adjusted to control the diameter size and the number of liposomes to maximize the encapsulation efficiency. For example, Schneider et al. revealed that itopride and Gd-DTPA encapsulation efficiency decreased from 50.6% to 34.2% and 50.1% to 43.2% when the liposome diameter decreased from 211 nm to 87 nm and 158 to 84 nm respectively. On the other hand, implementing FT 3 times increased encapsulation efficiency from 34.1 to 45.1% and 44.2 to 49.5% for itopride and GD-DTPA respectively, proving the previous claim by Ueno.38
High encapsulation efficiency may also be achieved via REV method which involves preparation of emulsion between lipid phase and small molecule therapeutic solution followed by evaporation of solvents and rehydration of the resulting residue to form small molecule therapeutic-encapsulated liposomes. This method is advantageous over other methods because it provides large internal aqueous space-to-lipid ratio which increases the average volume of small molecule therapeutic being encapsulated.54 REV results in large LUV liposomes which leads to as high as 60–65% in encapsulation efficiency.55 Compared to other methods such as FT, REV could encapsulate much better than FT for encapsulating identical small molecule therapeutics. For example, comparing encapsulation of 5-FU using FT and REV methods, REV method exhibited higher encapsulation efficiency but it was more variant in terms of the liposomal diameter.56 Furthermore, the pore size of the liposome formed via REV method is larger (0.2 μm) than FT-made liposomes (0.1 μm) which directly relates to the permeability. This means that REV-made liposome had easier release and higher leakage than FT made liposomes. A possible reason for inconsistent liposome diameter may be due to formation of multiple bilayer formation during emulsification using ultra-sonication, leading to formation of particularly larger liposome. When emulsification during sonication was not completed, lipid molecules may remain aggregated and therefore formed secondary bilayer around a pre-formed liposome.29
In another study, REV method was compared with usual TLH technique in the encapsulation efficiency of sodium cromoglycate and sumatriptan succinate. The results showed that for both small molecule therapeutics, REV method had significantly higher encapsulation efficiency.29,40 Depending on the arrangement of the lamellae, the encapsulation efficiency may also vary although they were prepared via same methods. Comparing amongst SUV, LUV and MLV, SUV had the lowest encapsulation efficiency while LUV had the highest. Considering this, Zhang et al. experimented to encapsulate salvinolic acid B using different encapsulation techniques such as TLH, ether injection and double emulsion, and the results proved that LUV based liposome prepared via REV technique had the highest encapsulation efficiency.57
Being different from previously mentioned methods, dehydration–rehydration vesicles (DRVs) method was used to prepare liposome under high small molecule therapeutic to lipid concentration ratio. The mixture of small molecule therapeutic and liposome was freeze dried and rehydrated before synthesis. This method was further enhanced with modification by preforming liposome from lipid dispersed in aqueous solution before adding hydrophilic small molecule therapeutics. The modified preparation technique known as dehydration and rehydration of preformed empty vesicles (DRPEVs) and it was performed using empty liposomes mixed with hydrophilic small molecule therapeutics. Empty liposome was re-formed during freeze drying and rehydration process which allows encapsulation of the surrounding small molecule therapeutics in a buffer solution. Experiments using penicillin, riboflavin, doxorubicin (DOX), deoxyfructo-serotonin and epidermal growth factor (EGF) revealed that the encapsulation of the small molecule therapeutic were heavily dependent on small molecule therapeutic properties and small molecule therapeutic to lipid ratio but not by lipid lamellar transition of the liquid crystalline.46 During the encapsulation of vancomycin small molecule therapeutic encapsulation, DRV technique produced much higher entrapment efficiency when compared to usual TLH technique and even when compared to active encapsulation technique for encapsulating ammonium sulphate.58
In review of recent techniques developed for liposome synthesis, several other liposome synthesis methods were discovered, studied and analysed.59 Novel techniques identified by this review includes: microfluids,60 supercritical fluids61,62 and electroformation,63–66 freeze drying of double emulsion,67 hydration of phospholipid deposited on nanostructured materials,68 curvature-tuning69 methods and many other.59 These novel techniques were noted to be advantageous as they can potentially encapsulate both polar and lipophilic small molecule therapeutics into liposomes.70,71
4.2. Methodology to increase active encapsulation efficiency
Active small molecule therapeutic encapsulation refers to the use of concentration gradients between membranes to encapsulate weakly ionised small therapeutic molecules. As known as remote loading, the active method of small molecule therapeutic loading usually offers a higher small molecule therapeutic encapsulation efficiency compared to the passive variant mentioned above in the review.D at pH 7 within the range between −2.5 and 2.0 and a pKa ≤ 11 for optimised remote small molecule therapeutic encapsulation. These conditions could be satisfied by amphipathic weak bases.75–77 Other than taking advantage of the pH gradient to drive the loading of DXR into liposomes, other techniques such as use of manganese gradients, sulphate gradients and citrate gradients have also be employed. Likewise, the free DXR base diffuses into the liposome where the small molecule therapeutics are modified and inhibits the membrane permeation. These results in the accumulation of the modified DXR within the liposome.78It is also important that amphipathic weak acids can also be encapsulated. Due to the permeability differences between the acetic acid molecule and phospholipid bilayer of the positively charged ion, the transmembrane acetate approach can be used for such a case as discussed by Clerc and Barenholz,79 where the pH transmembrane gradient was used as the driving force for assembly. Due to differences in the calcium acetate concentrations across the liposomal membrane, the internal pH value of the liposome increased and thus formed a pH transmembrane gradient since the pH out of the liposome barely charged. Release experiments also showed that the loaded liposomes were not irreversibly modified during the loading procedure. The small molecule therapeutics can still be released via disruption of the liposomal membrane or by disrupting the transmembrane gradient. The transmembrane calcium acetate gradient method combines a simple experimental protocol with high entrapment capacity and stability of the loaded liposomes. Hwang et al. further improved this method by using small molecule therapeutics such as diclofenac, insulin and fluorescein isothiocyanate-labeled insulin.74 In this study, a trapping efficiency of almost 100% was obtained for the encapsulation of diclofenac in liposomes via the calcium acetate approach, however, only about 1–8% trapping efficiency was observed for liposomes prepared from the conventional reverse-phase evaporation vesicle (REV) method.
Fig. 4 illustrates the small molecule therapeutic encapsulation process for DOX in a nano-liposome. The first small molecule therapeutic, Doxil® was developed in 1995 and approved by FDA. This was due to the development of a more successful remote method of small molecule therapeutic loading of the encapsulation efficiency of DXR by Barenholz and collaborators. Two other principles were responsible for the development of this small molecule therapeutic, namely the increased small molecule therapeutic circulation time and avoidance of the reticuloendothelial system (RES) of the liposome due to the use of PEGylated nano liposomes and also having the liposomal lipid bilayer composing of cholesterol and a high Tm (53 °C) phosphatidylcholine.22 This procedure involved a transmembrane ammonium sulphate gradient, where the concentration of ammonium sulphate within the liposome is higher than that of the medium. This acts as the driving force for this novel method of remote loading of the small molecule therapeutic. This method of loading into preformed nano-liposomes is also stable and efficient due to the ability of transferring of ammonium ions within the liposomes with weak bases located out of the liposome. When the small molecule therapeutic transited into the liposome, it precipitated as a sulphate salt. The stability of the ammonium sulphate ion gradient is due to the low permeability of the sulphate, which enables the stabilisation of the small molecule therapeutic accumulation over prolonged storage durations (>6 months) due to aggregation and gelation of the anthracycline sulphate salt, where DXR falls under the anthracycline class of small molecule therapeutics. One core advantage of deploying this ammonium sulphate gradient method for small molecule therapeutic encapsulation is that it is unnecessary for the liposomes to be produced within an acidic medium or to increase the alkalinity of the external aqueous medium.23
 | ||
| Fig. 4 Illustration for DOX encapsulation.22 | ||
In addition to the use of ammonium sulphate salts for active encapsulation of weak bases in liposomes, Fritze et al. in 2006 studied the remote loading process driven via a transmembrane ammonium phosphate gradient, from which the DXR was precipitated in the presence of sulphate and citrate ions as well.78 It showed that the loading efficiency of DXR was 98% in large unilamellar vesicles, which was composed of cholesterol/egg phosphatidylcholine (3/7 mol/mol) bearing a transmembrane phosphate gradient. Ammonium salts managed to achieve significantly higher encapsulation efficiencies (citrate 100%, phosphate 98%, sulphate 95%, acetate 77%) compared to loading of sodium salts (citrate 54%, phosphate 52%, sulphate 44%, acetate 16%). This difference in encapsulation efficiencies may be due to several reasons such as the capacity of the buffer, solubility of DXR in the different salt solutions, pH values and base counter flow. At physiological pH values, the small molecule therapeutic DXR was retained within the liposome and only be released when the pH value was decreased to 5.5, such as in acidic mediums (approximately 25% release at 25 °C or 30% at 37 °C within 2 h). Furthermore, the release rate was higher in cases of ammonium phosphate gradient applications compared to ammonium sulphate gradients. This newly developed small molecule therapeutic loading method however, is heavily dependent on pH for the liposomal small molecule therapeutic release. This pH-triggered release method may be applicable to treat tumour tissues. Metal ions may serve as another small molecule therapeutic loading method. A manganese gradient between MnSO4 and MnCl2 was used to compared with the previous methods of employing the ammonium sulphate and citrate gradients. This methodology was utilized to encapsulate a water soluble topotecan small molecule therapeutic while making use of the pH gradient. The methods enhanced small molecule therapeutic encapsulation, however, the liposomes prepared in the presence of MnCl2 and citrate demonstrated lower loading capacities of topotecan. Upon increase of topotecan's loading ratio, a reduced rate in small molecule therapeutic release was observed. This could be due to crystal formations of topotecan. The encapsulated topotecan precipitated as linear particles within the liposomes as seen on cryo-electron micrographs. Furthermore, the stability of the loaded liposomes was dependent on the presence of both the encapsulated sulphate as well as the pH gradient.80
Copper was also investigated as a metal ion to facilitate the liposomal encapsulation of mitoxantrone. The small molecule therapeutic can be efficiently loaded regardless of initial intraliposomal values and the types of anions used. In addition, the encapsulation efficiency decreased with an increased small molecule therapeutic-to-lipid ratio. Furthermore, during the small molecule therapeutic loading process, there is generation of a transmembrane pH gradient. The intraliposomal pH values may also affect the complexation between the Cu2+ ions with the mitoxantrone small molecule therapeutic. Due to the pH gradient, upon accumulation within the vesicles, some of the mitoxantrone became protonated and thus precipitated by the sulphate. From the results, for in vitro release, CuCl2-containing vesicles were able to release mitoxantrone at faster rates than CuSO4. In vivo studies on the other hand, revealed that CuCl2 formulations were more therapeutically active compared to CuSO4. This might be due to the differences of the state of the small molecule therapeutic physically when compounded with Cl2 or SO4.81
Another example of the metal ions is nickel, which can improve small molecule therapeutic encapsulation by forming a complex with mitoxantrone. In one study, Ni2+ was found to mediate effectively on the stable mitoxantrone small molecule therapeutic loading into large unilamellar vesicles. However, it was found that the small molecule therapeutic/Ni complex was membrane impermeable and thus stable. This is due to no burst effects observed during the in vitro release of the small molecule therapeutic. Furthermore, this transmembrane NiSO4 gradient was applied to facilitate effective mitoxantrone loading. The formulation prepared with fluid lipid also displayed a fast release rate of the small molecule therapeutics.82
Cern and his co-workers developed a mathematical model, also known as the Quantitative Structure Property Relationship (QSPR) model, in a study, aiming to assist the selection process of small molecule therapeutic encapsulation. This model included 60 small molecule therapeutics studied in 366 loading experiments, where the small molecule therapeutics were encapsulated in various incubation conditions such as the molecular physicochemical characteristics, phospholipid transition temperature, % of cholesterol used, membrane rigidity scale, the liposomal size, small molecule therapeutic-to-lipid molecular ratio, loading duration and temperature, type of gradient used, extent of pH gradient used, the salt concentration. This model correlated both chemical structural features with experimental conditions and thus enabled classification of small molecule therapeutic whether good or bad for use in remote loading. In this model, good candidates are those small molecule therapeutics which are able to achieve a high loading rate regardless of small molecule therapeutic-to-lipid ratios. This QSPR model can be used to identify small molecule therapeutics that are expected to have high remote loading capacities, while still optimising the design of the formulation experiments.83
4.3. Methodology to enhance target delivery of small molecule therapeutic-encapsulated liposomes
Delivery of encapsulated small molecule therapeutics can be performed via two different methods namely static (passive) delivery and remote (active) delivery. Both methods of delivery aim to transfer small molecule therapeutic molecule to designated cells in vivo, without significant leakage or biodegradation along the way. While both methods have respective pros and cons, it is important to understand and study on these delivery mechanisms which can help to maximize the effectiveness of the small molecule therapeutics upon application.Passive small molecule therapeutic delivery consists of largely two different mechanisms. First, delivery is achieved via EPR effect arose from highly permeable vasculature with poor excrete system at tumorous region of the body. This causes nanocarriers like liposomes to flow in via extravasation and deposited at these region, allowing small molecule therapeutic concentration to reach optimum.13,84,85 Increased phagocyte concentration via extravasation was also seen at region of inflammation, and this allows deposition of liposomes.86 Second, delivery can be achieved gradually if the liposomes are meant for immunogenic cells such as phagocytes with mono-nucleus. Generally, it is biological phenomenon for liposomes to accumulate phagocytes due to immunogenic effect of phagocytes. When the small molecule therapeutics are meant for diseases that affects the human immune system such as leukaemia and rheumatoid arthritis, liposomes will effectively reach the affected cells, therefore produce effective small molecule therapeutic delivery.87 Remote delivery of liposomes or active targeting involves attachment of receptor/recognition molecules at the surface of the liposome. Therefore, liposome can be recognized and enter specific target cells by endocytosis, and releasing the small molecule therapeutic. These surface molecules may be anti-bodies, antigens, short chain of functional polymer, glycogen, glycoproteins or ligands that may provide cell-specific recognitions. This delivery not only increases the delivery efficiency, but it also prevents internalization of liposomes at undesired cells which may lead to unwanted toxic effects.88 However, remote delivery is not perfect as its delivery efficiency is limited by conventional passive delivery rate via extrusion at tumour prone region. Even when the liposomes are designed to actively target cells, its rate of accumulating at desired region still depends on the rate it flows, causing most of the liposome to accumulate at region driven by EPR instead of cells where recognition is matched. Nevertheless, with remote surface, the liposomes will still be endocytosed more readily then passive liposomes, leading to enhanced small molecule therapeutic delivery intracellularly. Fig. 5 illustrates the different modes of internalisation of small molecule therapeutics that are delivered remotely by liposomes.11
 | ||
| Fig. 5 Modes of internalization of small molecule therapeutics delivered remotely via liposomes. | ||
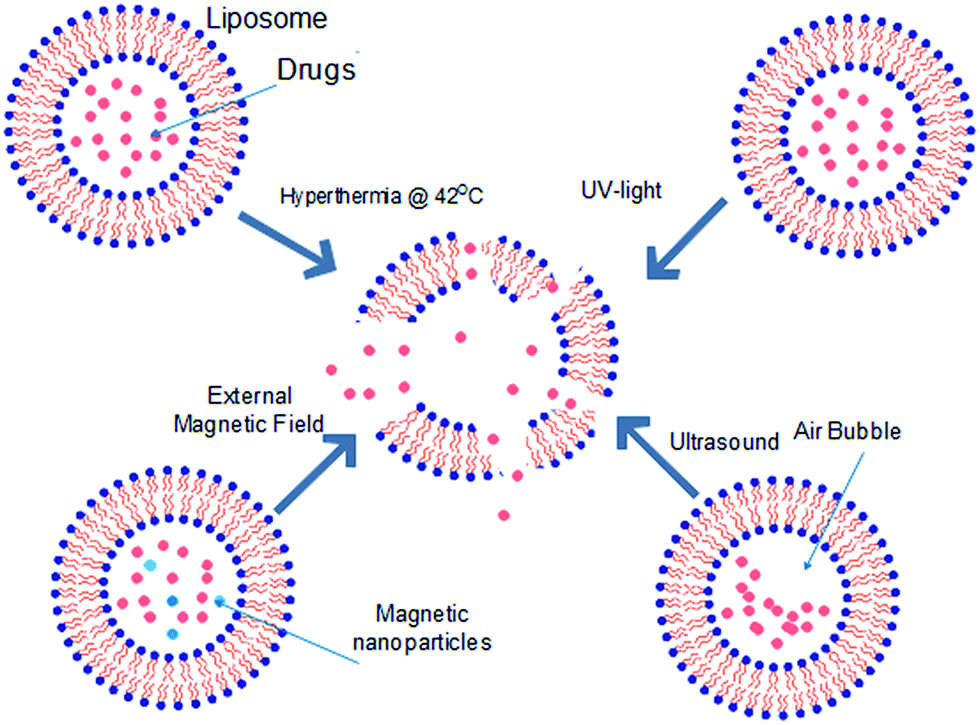
To remotely deliver small molecule therapeutics to targeted cells, liposomes may also need to be modified to release their content upon response to various stimuli. These stimuli may be triggers that are non-harmful to human and can be externally applied to induce response of liposomes in vivo. For example, liposomes may be thermosensitive (TSL),89–91 pH-sensitive (pHSL),92–94 echogenic (EL),95–97 magnetic resonance-sensitive (ML),98,99 light-sensitive (LSL)100 etc., depending on how their surfaces are modified as. Fig. 6 illustrates a summary of how these triggered release mechanisms of liposomes work. While these triggered delivery methods have already been actively used for lipophilic small molecule therapeutics, they were recently highlighted once again due to the potential to deliver polar molecules and small molecule therapeutics. For example, paclitaxel toxin which is lipophilic have been delivered successfully by liposome that responds to heat, magnet and ultrasound stimuli.101,102 According to Kopechek et al., comparison between delivery for polar and lipophilic small molecule therapeutics via echogenic liposome proved that polar small molecule therapeutics were released more readily than lipophilic small molecule therapeutics, showing that these stimuli-responsive delivery methods may be applicable to hydrophilic small molecule therapeutics with greater efficiency than delivery for lipophilic small molecule therapeutics.103 Hydrophilic small molecule therapeutics encapsulated at liposome remains at aqueous phase while lipophilic small molecule therapeutics exists at lipid phase. Upon exposure to external stimuli, liposomal membrane is broken and the contents released. Polar small molecule therapeutics will entirely be released along with the aqueous phase but some lipophilic small molecule therapeutics may interact with liposome fragments (phospholipid bilayers fragments) at the hydrophobic region, reducing the amount of small molecule therapeutic being released. Therefore, triggered delivery methods may better suit hydrophilic small molecule therapeutics. Additionally, highly polar small molecule therapeutics often experiences low penetration through tumour cells, triggered delivery methods such as pH-sensitive liposome can be used to encapsulate these small molecule therapeutics prior to delivery. This will increase the penetration efficiency and therefore increase bioavailability at the tumour region.104 Liposomes may also be engineered to attain specific functions to enhance delivery to targeted cells or intracellular organelles. According to Shelley et al. liposomal nanosystem could be used in targeted delivery of small molecule therapeutics to mitochondria of cells, to cure mitochondrial dysfunction such as respiration stress, DNA mutation due to viral infection such as Alzheimer's disease and diabetes.105 Occasionally, small molecule therapeutic delivery efficiency and bio-distribution were further improved with application of two or more different modes of delivery. For example, engineering of liposomal surface with functional polymer or antibodies allowed smooth endocytosis of the content, releasing the small molecule therapeutics into the cell. However, the internalized small molecule therapeutics targeted by lysosome were rapidly decomposed, leading to failure in expression of necessary effects. Thus, a secondary measure has to be imposed to the small molecule therapeutics to ensure that small molecule therapeutics are not degraded by lysosome and only be released at cytoplasm.106
 | ||
| Fig. 6 Illustrative summary for different modes of triggered release. | ||
In this section, the passive and remote delivery mechanisms for polar and mildly ionized small molecule therapeutic molecules encapsulated in liposomes are summarized, and the constituents, the triggering mechanisms and experimental results are discussed.
The use of liposomes as detection agents may be also potentially effective in diagnosis of cancerous tumours. Conventionally, tumours could only be detected by traditional imaging system when the size of the tumours have increased up to 1 cm in diameter. However, using liposomes as contrasting agents, tumours as small as 2–3 mm in diameter can be detected. In these cases, the EPR effect causes nanocarriers like liposomes to accumulate at the cancerous region via extravasation.118–121 EPR effect is expressed at tumours of such small size because blood vessel expansion via neoangiogenesis which have started to supply necessary energy source and oxygen to tumorous cells, and hence liposome could be effective in detecting small tumours in the body.118,122,123
 | ||
| Fig. 7 Structural illustration summary for functionalization of liposome.124 | ||
The liposomes can be modified at its surface to possess specific functional groups to bind with the corresponding sites of the target cell for small molecule therapeutic administration. The inclusion of a peptide function group which is attached to integrin is found to facilitate cell penetration of the administered small molecule therapeutic.125,126 One such example includes the use of octa-arginine, which functions as a cell-penetrating peptide, where the octa-arginine is coupled to a polyethylene glycol-dioleoyl phosphatidylethanolamine amphiphilic copolymer which is then attached to a liposome. These liposomes increased the suppression of tumour growth in vivo and increased cytotoxic activity in vitro as compared with the addition of octa-arginine.116 HIV TAT (TAT is a 86 amino-acid protein) protein, using the 36 amino-acid domain/sequence, is able to achieve a very fast method of small molecule therapeutic administering to the target cell.127,128 There are other cell-penetrating peptides such as penetratin (Antp) and VP22.
Another group of added peptides include those which are able to bind to ligands found on the targeted cell surface. Due to the restricted exhibition of siglecs (sialic acid-binding immunoglobulin-like lectin) on one if not a few leukocyte cell types, siglecs are increasingly popular as a candidate for cell-targeted therapy. CD22 is a molecule of the siglec family which is found on B lymphocytes. There is a type of white blood cells which is able to recognise glycans of glycoproteins as ligands. Ligand-coupled liposomes can be readily bound of CD22 and thus quickly absorbed into the target cell via endocytosis.129 Other methods includes targeting the overly expressed hyaluronan receptor CD44 together with the RHAMM (receptor for hyaluronan mediated motility) protein in cancer cells by targeting specifically these naturally-occurring high-Mr hyaluronan.130
The second approach includes targeting of folate receptors (FR) via the coating of derivatives of folic acids or folates on the liposomes due to the overexpression of FRs in human cancer cells.131 Using DOX as an example of a small molecule therapeutic, folate-polymer-coated liposomes are used as a delivery vehicle. The liposomes were specifically coated with folate-poly(L-lysine) (F-PLL) conjugates which had a folate modification degree of 16.7 mol% on the epsilon amino groups of the PLL. DOX-loaded anionic liposomes were then coated with the synthesised F-PLL before use. Cytotoxicity of F-PLL-coated liposomal DXR was observed to be double of that of without folate modification, suggesting the occurrence of FR-mediated endocytosis.132
Coating of antibodies onto the liposomes is another approach. This method can be done by either coating of phospholipid head group or of the end of the polyethylene glycol polymer. Such as in breast cancer treatment, the overexpression of heparin-binding epidermal growth factor-like growth factor (HB-EGF) in cancer cells was adopted to develop a targeted small molecule therapeutic delivery system. The antibodies-coated liposomes (immunoliposomes) were able to associate significantly with Vero-H cells, which are one of the most widely used mammalian cell lines with overly expressed HB-EGF compared to wild-type Vero cells. On the other hand, liposomes without any coating were able to bind to neither cells.88
With employment of fusogenic lipids and peptides, endosomal escape was achieved. Fusogenic lipids and peptides destabilise the endosomal membrane with a conformational transition at low pH.137,138 A typical example is liposomes consisting of dioleylphosphoethanolamine (DOPE) and cholesterylhemisuccinate (CHEMS) where at mildly acidic condition, liposomes with this composition are unstable and undergo fusion with membranes of endosome or lysosome, releasing the encapsulated content.93,139 The reason for this phenomenon is due to the chemical structure of the composition whereby conical shape of DOPE has only a small hydrophilic head with long lipophilic tail, while CHEMS has an inverted conical shape at physiological pH. However, as CHEMS possesses much larger portion of hydrophilic carboxylic groups that will undergo hydration at low pH, the conformation will change from conical structure, leading to disruption in the macrostructure from lamellar to hexagonal phase of liposomal membrane.140,141 For DOPE with lipid compound, the lipid will get protonated in low pH and this can lead to a decreased propensity for water binding, hence resulting in lipid dehydration. Since lipid hydration creates a steric barrier inhibiting at short distances, the lipid dehydration can induce the disorganization of the membrane and small molecule therapeutic release into cytosol.142
Oligopeptide chains that induces membrane fusion, also known as fusogenic peptides, have also been actively studied to induce cytoplasmic delivery.138 Previously, a glutamic acid-based zwitterionic lipid, 1,5-dihexadecyl N,N-diglutamyl-lysyl-L-glutamate, was used as a pH responsive component for liposome system to test for target delivery of DOX into cytoplasm. The peptide chain exhibited charge inversion from negative to positive at decreasing pH, which could cause membrane disruption and release the contained small molecule therapeutic into cytoplasm. Cell culture results showed an enhanced release of small molecule therapeutics and anti-tumour effects compared with the conventional liposome.143 In another study, Gomez et al. used GALA peptides to attach to the surface of prostate specific membrane antigen (PSMA)-targeting liposome for DOX delivery.144 Upon PSMA induced endocytosis, endosome's acid environment led to change in oligopeptide conformation and further caused endosomal membrane disruption and release of the encapsulated small molecule therapeutic into the cytoplasm of tumour endothelial cells.
In addition to the anti-cancer effects, the studies of using pHSL for cytoplasmic small molecule therapeutic delivery in cells affected by other diseases are also receiving increasing attention. One typical example is DOPE/CHEMS-based pHSL encapsulated etanidazone which resulted in a fast and large amount of small molecule therapeutic delivery into murine J774 macrophages (from 0% for the free small molecule therapeutic to 72% for pHSL), being phagocytosed by an uninfected and trypanosoma cruzi-infected cell, in addition to causing a significant reduction of parasitemia in infected mice.93 In addition, pHSLs are also capable of acting as imaging probe for inflammation sites where physiological acidity of the region is abnormally high.94,145,146 For example, pHSLs formulated with the fusogenic phospholipid1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) and the membrane stabiliser D-α-tocopherol-hemisuccinate with magnetic resonance imaging (MRI) showed fast and fully released at pH 5.5 in vitro after incubation with J774 macrophages.92
In another study, antibiotic ceftizoxime (SpHL-99mTc-CF) was encapsulated into DOPE/CHEMS-based pHSL to detect and alleviate osteomyelitis. Mice labelled with technetium-99m with induced osteomyelitis by Staphylococcus aureus foci at the left tibia was used in the in vivo study. For healthy animals, the radioactivity level was identical for both left and right tibias, while the infected left tibia showed the radioactivity level were 1.5 fold greater than the right tibia without inflammation. Thus, pHSL loaded with ceftizoxime showed potential for the diagnosis of bone infections.94 Other than having compositional pH sensitivity at liposomes, integration of some pH sensitive poly/oligomers such as poly(styrene-co-maleic acid) (SMA) were used. SMA showed conformational transition from a charged extended structure to an uncharged globule under its pK1 value, to grant a pH sensitive property to liposomes. Synthesis of pHSL was also identified to be more stable under physiological condition than conventional liposome. It showed that no haemolytic activity was detected at physiological pH, while enhanced programmed cell death to colon cancer HT-29 cells was maintained.147
Another method for small molecule therapeutic delivery using liposome is to skip endocytosis. In this mechanism, liposomes fuse with membrane of tumour cell directly, and release the encapsulated small molecule therapeutic directly to induce apoptosis or anti-cancer effect. This mechanism is possible as tumour cells have lower pH like inflammatory regions. The lower pH in tumour cells is caused by increased cell activity that leads to increased rate of glycolysis, producing more lactic acid and ADP at the hypoxic region of tumour.148,149 Direct cytosolic delivery was achieved by incorporation of pH-low insertion peptides (pHLIPs).150 Above pH 7, pHLIPs are in equilibrium between aqueous phase and surface of lipid phase. However, at lower pH, pHLIPs forms a helix that shifts the equilibrium. Therefore in usual healthy tissue with pH 7.0, pHLIP will only bind to cell surface, whereas in inflamed and infected tissue, usually having an acidic environment, pHLIP will insert into the cells.151,152 When pHLIPs is within the liposomal structure, it fuses these two membranes, increasing cellular uptake and releasing the content into the cytosol.153–156 The study revealed that the affinity of pHLIP to cells are higher at lower pH,153,154 and pHLIP liposome indeed released its internal content directly into cytosol. pHLIPs can potentially be employed to improve the cellular uptake and intracellular release of cell-impermeable molecules, such as hydrophilic small molecule therapeutics.152
pHSL was also functionalized via addition of antibodies and other ligands to increase cell selectivity. One study revealed that DOX-loaded immunoliposomes containing the antibody mAb 2C5 (multiple tumour detector), cell penetrating TAT moieties (enhanced cellular uptake) and PE (PEG2k-Hz-PE) lipid as pH-sensitive shielding barriers could achieve cytoplasmic delivery of small molecule therapeutics. Upon reaching tumour site via extravasation, the acidity caused hydrolysis of PEG chain, which would not occur at normal physiological condition. TAT moieties were shielded by long PEG chains and TAT extended outward to increase transmembrane diffusion, enhancing the interaction with tumour cell as well as the cytotoxicity of DOX against B16-F10, HeLa and MCF-7 cells after incubation at acidic pH.106 Modified surface-targeting liposome with dihene-icosanoyl phosphatidylcholine and distearoyl phosphatidyl serine for targeting delivery of DOX was reported. In this design, the targeting ligand remained unexposed during blood circulation, while it was exposed to surface when the liposome reached tumourous region where pH is reduced, which increased sensitivity to target to kill cancerous cells. From the experiment, the delivery of DOX at pH 6.5 resulted in increase in targeting from 41 to 93% while rate of cell destruction increased from 23 to 71%.157
Gemcitabine hydrochloride encapsulated liposomes were prepared using DOPE/CHEMS-based pHSLs with epidermal growth factor receptor (EGFR) antibody as ligand, to target non-small lung cancer cells. It showed that the cell mass of tumour treated with EGFR-pHSL was considerably less than those with non-targeted pHSL, revealing that EGFR-pHSL leads to sharp reduction in the tumorous cell proliferation and increases the chance of cell programmed death.158 When anti-CD33 monoclonal antibody was attached to cytosine arabinoside (Ara-C)-loaded pH-sensitive immunoliposomes, pH-sensitive copolymer poly(N-isopropylacrylamide-co-acrylamide) [P(NIPAM-co-MAA)] of the liposome actively targeted leukaemia cells. Acidic pH of the region caused the copolymer to shrink, creating a curvature in the bilayer plane, leading to defect in the liposomal membrane and releasing the encapsulated small molecule therapeutic.159–163 Further analysis revealed that pH sensitive immunoliposomes with anti-CD33 antibody were effectively engulfed by leukemia cells (with higher cytotoxicity against HL60 Cells), showing that the presence of NIPAM could lead to release of Ara-C in the endosome.163 Liu et al. synthesized DOX and verapamil-co-loaded pHSL which was made from compound malachite green carbinol, circumvented the P-glycoprotein-mediated small molecule therapeutic resistance in Her-2(+) breast cancer through addition of Her-2 antibody.164 The results showed that pHSL was able to penetrate MCF-7 Her-2(+) DOX resistant cells efficiently. With higher cytosol DOX deposition and greater release at tumour site, the IC50 in DOX-resistant MCF-7 was reduced by 6-fold in the tumour bearing mice, consequently leading to higher tumour suppression and decreased toxicity. For treatment of solid Ehrlich tumours in mice, 159Gd-DTPA-BMA (gadodiamide) loaded DOPE/CHEMS-based pHSL and folate-pHSL were used, which showed 4 and 14 times higher penetration than free small molecule therapeutic respectively.165 However, in spite of higher penetration for folate-pHSL, efficiency was not proportionally higher.166
4.3.4.1. Magnetic liposome (MLs). Magnetic nanoparticles made up of iron oxide molecules of either maghemite (γ-Fe2O3) or magnetite (Fe3O4) can also be encapsulated to obtain magnetic liposomes (MLs). MLs enable triggered delivery of the small molecule therapeutics with the use of an external magnetic field. Saiyed et al.167 developed magnetic azidothymidine 5′-triphosphate liposomes and demonstrated the transmigration of the encapsulation small molecule therapeutics could cross an in vitro blood–brain barrier model with the use of an external magnetic field.
MLs have several potential uses such as MRI contrasting agents for diagnostic purposes,168–170 in cancer treatment as temperature mediators in hyperthermia treatment therapies using an external alternating magnetic field171,172 as well as its ability to compound with various small molecule therapeutics for triggered release to improve effectiveness and personalisation of the treatment. In addition, the use of magnetic nanoparticles in liposomes can reduce the level of toxicity while increase their effectiveness as MRI contrasting agents.173,174
The general direction for the use of magnetic small molecule therapeutics delivery systems, specifically for cancer therapy, is to enhance local effectiveness as well as to reduce potential side effects of the treatment.167 Furthermore, such systems are potentially efficient methods in the making of imaging and therapy agents to cross the blood brain barriers, thus attaining the required levels to diagnose and treat brain cancer cells.167,175 There are two general categories of MLs. The “classical ML”, uses a 15 nm iron oxide nanoparticle core that is surrounded by a phospholipid bilayer on its surface. The nanoparticle core is fully filled by the iron oxide particle and thus lacks the presence of an aqueous medium. This “classical” type of ML has been ideally used for MRI purposes. However, due to its nature, it is unable to encapsulate hydrophilic small molecule therapeutics. The latter type on the other hand comprises of larger lipid vesicles, between 100 and 500 nm, encompassing several iron oxide nanoparticles of approximately 1–10 nm, that are randomly dispersed within an aqueous core. This in turn facilitates the hydrophilic small molecule therapeutics to be co-encapsulated.176,177
In one example, Saiyed et al.167 developed MLs encapsulating AZTTP. This design was attempted to increase transmigration across an in vitro blood brain barrier model, using of HIV-infected peripheral blood mononuclear cells coupled with an external magnetic stimulus to treat neuroAIDS. It showed that MLs managed to improve the permeability of AZTTP by 3 times compared to the free AZTTP. Furthermore, the MLs were successfully absorbed by monocytes and the magnetised monocytes were responsive to external magnetic field and led to enhanced transendothelial migration.167
Generally, small molecule therapeutics release in MLs are controlled by magnetic fluid hyperthermia (MFH), through the application of a high-frequency alternating magnetic field (HF-AMF), such as for DXR loaded MLs.99 This enables a localised heating of the iron oxide nanoparticles and further causes thermal ablation of the targeted cells while preventing consequential damage to other tissues. This application of the HF-AMF damages cancer cells irreversibly upon reaching elevated temperatures ranging from 42 to 45 °C through heating178,179 which eventually leads to necrosis and apoptosis.180 For example, 5-FU loaded MLs possessed high in vitro blood biocompatibility as well as small molecule therapeutic releasing ability through the employing of magnetic gradients. In addition, the system exhibited small molecule therapeutic release triggered by hyperthermia, suggesting future applications for antitumour therapy.179 A similar study of MLs using calcein as small molecule therapeutic model also resulted in the releasing of the encapsulated small molecule therapeutic via both triggered agents, in which the magnetocaloric effect caused liposomal phase transitions as well as the magnetic-impelled motions and thus improved permeability of the bilayer.181
The release of encapsulated small molecule therapeutics from MLs through the use of a low-frequency alternating magnetic field (LF-AMF) in the absence of additional heat was also investigated. In a study, cobalt ferrite (CoFe2O4) nanoparticles and carboxyfluorescein were used as hydrophilic model compounds and loaded into MLs. These MLs were then exposed to LF-AMF to observe the subsequent effects. The application of the magnetic field strongly increased the permeability of the MLs membranes and triggered the release of carboxyfluorescein followed zero-order kinetics. These results indicated the rising potential of this system as an effective method for future applications of small molecule therapeutics delivery.98
MLs containing hydroxyethyl starch-coated iron oxide nanoparticles have been developed. This aims to deliver fasudil to pulmonary vasculature for pulmonary arterial hypertension treatments through intrathecal administration. The toxic side effects of using magnetites were reduced due to the use of biodegradable and biocompatible starch coatings in the liposome. Furthermore, through the use of a magnetic field, the results showed a 3 time increment of the cellular uptake into pulmonary arterial smooth muscle cells (PASMC). On the other hand, for both plain liposomes and MLs which functioned in the absence of a magnetic field, there is a general reduction of the proliferation of 5-HT-induced PASMC.182 The surface of the MLs were also functionalised for targeted therapy. In one study, DXR and anionic superparamagnetic iron oxide nanoparticles were formulated into cationic MLs containing folate as the targeting group. Upon heating at radio frequencies, from an alternating electromagnetic field, three times increment of the DXR small molecule therapeutic release were observed. Furthermore, there was significant surface binding as well as high cellular uptake in the FR-overexpressed cervical cancer cells. However, there was no uptake observed for human breast carcinoma cells which expressed lower FR.183 Arginyl-glycyl-aspartic-acid (RGD) is a peptide, composed of three acids, namely the L-arginine, glycine, and L-aspartic acid. RGD-coated MLs, loaded with sodium diclofenac, targeting cerebral inflammatory sites demonstrated a 9.1 times increase in small molecule therapeutic brain levels compared to the free small molecule therapeutic, a 6.62 times higher levels than non-magnetic RGD-coated liposomes and also 1.5 times higher levels compared to uncoated MLs under the influence of a magnetic field. However, these RGD-coated MLs were avoided by macrophages of the liver tissues, whereas they were only taken up by mobile monocytes and neutrophils selectively.184 Instead, a cyclical RGD-peptide was further developed to bind onto the surface of MLs containing gadolinium diethylenetriamine penta-acetic acid (Gd-DTPA). Due to the influence of RGD-peptide, the affinity of the ML with respect to the tumour cells increased. Furthermore, it also enabled the MLs to specifically target tumours in vivo, thus improving the magnetic resonance (MR) signal in mice bearing A549 tumours.185 The targeted delivery and cellular uptake of folate-coated DXR-TSML were improved by taking into account several factors such as the targeted delivery systems, temperature sensitivity and magnetic properties. This formulation showed sensitivity towards temperature, strong responsiveness towards magnetic fields as well as increased tumour cell uptake as mediated by FR. This is superior to non-magnetic folate-targeted liposomes and the commercial stealth liposomal formulation Caelyx®. The DXR cytotoxic effects were also increased due to the integration of both biological and physical small molecule therapeutic targeting.186
4.3.4.2. Thermosensitive liposomes (TSL). In tumour sites where local hyperthermia of the patient is employed to facilitate small molecule therapeutic release, Yatvin et al.187 and Weinstein et al.188 developed thermosensitive liposomes (TSLs) for this matter. This method employed a system whereby the lipid membrane exhibited a phase transition temperature (Tm) slightly above the physiological temperatures of average humans. Hyperthermia, as a medical condition, seems to be a more effective small molecule therapeutic release triggering method for tumour treatment. These regions exhibit increased blood flow and circulation while increase permeability at the microvascular level in addition to appear to be cytotoxic in nature.88 Another benefit of using hyperthermia-based small molecule therapeutic delivery is that it is not entirely reliant on the EPR effect. This is due to TSLs being administered only during hyperthermia treatments. The small molecule therapeutics are then quickly released into the microvessels of the tumour. After which, the surrounding cells, regardless of vasculature nature (leaky or not) will experience a rapid uptake of the small molecule therapeutics. In addition, the small molecule therapeutics are delivered to the tumours in free forms. This improves the bioavailability and tumour penetration rates.189–192 Encapsulation of the small molecule therapeutics into TSLs should be performed at about 37 °C, whilst released within the range of 39–42 °C. This is mainly due to intratumoral temperatures not exceeding 42 °C during the administration of the small molecule therapeutics.89,193 One major setback for the use of hyperthermia as a small molecule therapeutics release trigger is that it is ineffective regarding treatment of metastasis owing to the characteristic of the TSLs, where a precise understanding of the tumour location is to be known.194
Several studies described TSLs that contain the temperature sensitive phospholipid DPPC,89,90,195 which has a Tm of about 41.9 °C.196 Another variant of TSLs contains DSPC, which is another lipid bearing similar properties with DPPC.88 Generally, the thermosensitive phospholipid is closely associated with lysolipids such as monopalmitoyl phosphatidylcholine (MPPC)89,197 and monostearyl phosphatidyl-choline (MSPC),90,91 thus a compound such as lysolipid containing thermosensitive liposomes (LTSLs). The presence of the lysolipid on the TSLs strongly enhances the effectiveness and efficiency of small molecule therapeutics release, possibly due to enhancements in the permeability of the liposomal membrane.196
Based on DPPC and MPPC, vinorelbine bitartrate-LTSLs were administrated to mice with lung tumours. The results showed that tumour inhibition was significantly higher than their non-LTSL and free small molecule therapeutic counterparts.89 Similarly, another study, specifically on epirubicin (EPI) loaded within DPPC/MSPC-based LTSLs for rats, resulted in a higher circulation time than their non-LTSL and free small molecule therapeutic counterparts. The average tumour inhibitory rate in mice with the Lewis lung carcinoma was 60% for LTSLs, whereas both non-LTSL and free small molecule therapeutics were 40%.90 DXR-containing LTSLs based on DPPS and MSPC were further evaluated in mice bearing C6 glioma. After experiencing hyperthermia at 42 °C, the maximum brain concentration of DXR was achieved for the LTSLs, notably 6.4 times more than free small molecule therapeutic and 3.7 times more for the non-LTSL cases. Furthermore, the survival windows were prolonged much more for the LTSLs.195
For the association of therapeutic as well as diagnostic agents, both TSLs and LTSLs were studied. De Smet et al.197 managed to observe from two separate temperature formulations, namely, DPPC-based TSL and DPPC/MPPC-based LTSL, its effects on the release of DXR, which was jointly encapsulated with the MRI probe [Gd(HPDO3A)(H2O)]. The DXR release rate was not affected by the presence of the contrasting agent. Furthermore, LTSL resulted a faster release of DXR at 40 °C than TSL, however, LTSL showed a higher leakage of DXR at 37 °C.
Li et al. prepared DXR loaded in 2 different types of liposomes, namely the DPPC/DSPC based TSL and DPPC/MSPC based LTSL.91 After a single dosage of the small molecule therapeutic followed with a mild hyperthermia induction, the mice transplanted with subcutaneous human BLM melanoma showed an improvement for tumour suppression for TSL compared to LTSL. It showed that TSL caused a suppression of tumour growth of over 16 days whereas LTSL showed a delay of merely 8 days, half of that of TSL. In addition, 6 out of 9 mice treated with the TSL survived, whereas only 2 out of the 9 treated with LTSL managed to survive after 26 days of post treatment. In another study, Li et al. reported that the small molecule therapeutic retention at physiological temperature was increased whilst still hindered the release of DXR.198 This was made through the modifications of the quantitative composition of the TSL which was developed to achieve a higher value of Tm. In this study, a two-step mild hyperthermia treatment method was employed with its aim of heating up the tumour site with more precision to avoid heating of the surrounding areas to treat deeply seated tumours or larger ones. This reduced small molecule therapeutic redistribution towards the other surrounding tissues. This may also be due to the characteristic of the TSL, being known to have a slower small molecule therapeutic release rate. For this method, the local area was firstly heated to an elevated temperature of 41 °C, after which the formulation of TSK was administered immediately after the occurrence of maximum extravasation and tumour accumulation. Finally, the area of the tissue containing the tumour was further heated up again to 42 °C to induce a local DXR small molecule therapeutic release.
DXR-loaded TSLs, containing mainly DPPS (96%) together with a little amount (4 mol%) of the surfactant Brij78 (polyoxyethylene (∼20 units, MW ∼ 880) stearyl ether) to substitute the lysolipid, were used to compare with a DPPC/MPPC/DSPE-PEG(2000)-based LTSL composition that is used in clinical trials (ThermoDox®).220 This new formulation showed a 1.4 times increase for small molecule therapeutic delivery at the tumour sites when compared to the LTSL formulation. Furthermore, it also exhibited increased tumour suppression and toxicity reduction.199 In a subsequent study, DOX and Gd-DTPA were jointly loaded, this was to analyse the DOX delivery effectiveness and possibly predict the potential for future therapies with such formulations in mice. The results indicated great potential of this system in various fields, such as the ability to quantify the amount of small molecule therapeutic delivery after treatment, evaluate prognosis and also a possibility for future treatment techniques at an personalised level.200 The pharmacokinetics also showed that a two times improvement in the small molecule therapeutic delivery to the heated tumours with the first LTSL, in addition to showing total inhibition of tumour growth, yet very little toxicity.189
In order to improve the thermosensitivity of DXR loaded TSL, DPPC was compounded to an elastin-like polypeptide. cRGD was used to modify the TSL surface to enable the binding to αvβ3 integrin that is overly expressed in angiogenic vasculature and tumour cells. After the induction of hyperthermia, 75% of the DXR was released at 42 °C and an increased release rate of 83% was further observed at 45 °C. The TSLs also showed combinational effect in the control of U87MG cells in cRGD targeting and thermal triggered release. Furthermore, the in vivo accumulation at the tumour site was 5 times more than non-modified TSLs.201
Thermosensitivity of the LTSL was also improved with the association of thermosensitive phospholipids with several natural thermosensitive polymers. An interesting property of these polymers is that they possess a lower critical soluble temperature (LCST), where below that temperature, these polymers are soluble in water, whereas above this temperature they become insoluble.202–204 This behaviour is due to dehydration and aggregation upon heating above the cloud point.205 By incorporating a polymer with a LCST, one could allow to adjust the stability of the liposomes in the target tissue through the employing of precipitation.161,206
Kono et al. developed a DXR-TSL which comprises of a PNIPAM derivative and poly(EOEOVE)-OD4 rather than using a thermosensitive phospholipid.88,207 This compound was obtained from a reaction process between octadecyl vinyl ether and poly[2-(2-ethoxy)ethoxyethyl vinyl ether]. After tumour heating at 45 °C, the TSLs were administrated into mice with colon carcinoma cells. Results showed that tumour growth was significantly suppressed during 6–12 hours after injection. However, only a minute suppressive effect was observed upon application of a mild heat of 37 °C. These TSLs were further jointly loaded with another compound G3-DL-DOTA-Gd. MRI imagery results showed that there was increasing accumulation of the small molecule therapeutic concentrations in the tumour until 8 hours after injection, before subsequently levelling out, indicating an impactful effect in tumour growth suppression.208 A copolymer comprising of PNIPAM together with the pH-sensitive propylacrylic acid was used, harnessing the temperature and pH dependence for the loading of DXR small molecule therapeutic. Results showed that such a method displayed a 120 times reduction in the thermal dosage levels necessary for releasing of the small molecule therapeutic at neutral pH environments. Furthermore, 1400 times reduction was observed at acidic conditions when compared to the conventional TSL methods. These results demonstrated the effective reduction of necrosis risks of healthier tissues.206
Another type of TSL was reported by using N-(2-hydroxypropyl)methacrylamide mono lactate and dilactate and formulated together with a range of various cloud points of a specific cholesterol anchor (Chol-pHPMAlac) for small molecule therapeutic DXR delivery. For such a system, the transition temperatures were adjusted by the molecular weight of the polymer chains as well as the density of the grafts to further control the small molecule therapeutic release. It showed that DXR was released at high temperatures as induced by high intensity ultrasound pulses. The results showed a good relationship between the effectiveness of the release of the small molecule therapeutics with the cloud points of the Chol-anchored polymers.207
4.3.4.3. Echogenic liposome (ELs). Due to non-invasive characteristics of echogenic liposomes, ultrasound-triggered release methods have been widely studied. Another advantage of such a method includes the ability to enter the body whilst increasing permeability across cell membranes and blood-tissue barriers.97 The driving force behind echogenic triggered release methods is mainly due to ELs' ability to react to ultrasound stimuli for small molecule therapeutic release, which is caused by trapped air within the liposomes. It is assumed that the behaviour of any hydrophobic small molecule therapeutics are similar to that of the encapsulated air, thus trapped within the monolayers of the liposomal lipid membrane, or as an air bubble layer encapsulating the aqueous core.96
Initial studies with ELs involved these vesicles as contrast agents.210,211 However, ELs are being further studied for use as small molecule therapeutics delivery mechanisms. In the presence of a low-intensity ultrasound, it is possible that the encapsulated content can be released from the liposomes, be it for small molecule therapeutic delivery purposes or for use as contrasting agents. This is due to the capability of ELs co-encapsulating both a gas as well as a hydrophilic small molecule therapeutic within the liposome.96 The release rates of the encapsulated small molecule therapeutic can be mediated via the control of the frequency of the ultrasonic pulses. A sustained release can be achieved using a consistent series of pulses at low amplitude. On the other hand, for a discrete amount of small molecule therapeutic release, a single ultrasound pulse of high amplitude can be employed. Small molecule therapeutic transport into the walls of arteries may be enhanced due to the air bubble generation in this ultrasound-driven mechanism. This leads to possibly an increase in permeability of the membrane whilst improving cellular transfection, where there is deliberate introduction of new species into the cells.212,213 The ultrasound-sensitivity of ELs is mainly due to an air pocket bubble expansion within the ELs, which is caused by the collision of the negative ultrasound waves. If this expansion of the air bubble is sufficiently large enough to overcome the elasticity limit of the membrane surface, the bilayer is overcome and the contents, where in this case the small molecule therapeutic, is released in this process.96 Kee et al.95 developed papaverine hydrochloride loaded EL and studied their ultrasound-mediated intravascular small molecule therapeutic release. This small molecule therapeutic, however, requires a high vascular concentration within targeted vascular bed. On the other hand, severe side effects were observed if the concentrations within the systemic circulation were to exceed a threshold, and thus the concentration must be controlled at low levels. In this study, papaverine kept its phosphodiesterase inhibitory activity upon encapsulation within the ELs, showing that ultrasound-triggered small molecule therapeutic release methods may be a promising system for a safer employment of this small molecule therapeutic specifically.
Furthermore, another method of using echogenic liposomes including the containment of emulsion, as known as emulsion-containing echogenic liposomes (eELs) was used to encapsulate a nanodroplet of perfluoropentane (PCF5) within the aqueous core of DPPC-based liposome. The liposome was prepared to test for a low-intensity ultrasound-mediated delivery of the DXR small molecule therapeutic. The results showed that eELs was able to release 80% of the DXR content upon exposure to ultrasound, whereas ELs without emulsion were only able to release 50% of the DXR content. Furthermore, there was a significant reduction of the proliferation of human cervical cancer (HeLa) when eEL was used together with the use of ultrasound. Further study demonstrated that EL-DXR carriers were biologically compatible to live cells in vitro and may have the potential for use as an effective small molecule therapeutic carrier for ultrasound-triggered small molecule therapeutic delivery mechanisms.95
4.3.4.4. Light-sensitive liposomes (LSLs). Optical diagnosis and treatment methods are becoming increasingly popular these days due to its high spatial imaging resolution, ability for low-invasive treatments as well as the possibility of localising treatments of several diseases such as cancer, inflammation and skin injuries.214 Amongst ultraviolet light, visible and near-infrared light, the near-infrared light is able to penetrate into the deepest tissues. This makes the near-infrared light highly desirable for cancer treatment. Photodynamic therapy has become a widely used method for the superficial tumour treatment.215,216 Photosensitising agents, such as porphyrin derivatives, chlorins, phthalocyanines and porphycenes can generate species of oxygen radicals upon the exposition of light, which are used to target malignant cells.138,217,218 One potent, widely used photosensitising agent is the lipophilic compound meta-tetra(hydroxyphenyl)chlorin (mTHPC or temoporfin). For palliative treatment of advanced squamous cell carcinoma (SCC) of the head and the neck, a formulation comprising of ethanol, propylene glycol together with mTHPC (Foscam®) has been approved for use. There are two liposomal formulations available for use as well, Foslip®, which consists of mTHPC trapped in conventional liposomes while Fospeg® uses PEGylated liposomes.219
Liposomes were previously formulated using light-sensitive lipids to trigger photo-induced structural changes of the membrane of the liposome. Yavlovich et al.220 and Romanowski221 demonstrated that several mechanisms such as photoisomerisation, photocleavage and photopolymerisation could cause liposome destruction and consequently release the small molecule therapeutic contents. Based on this concept, it is possible to load hydrophilic small molecule therapeutics into the liposomes which are light triggered. For example, a liposomal formulation comprising of DPPC, photopolymerizable diacetylene phospholipid (DC(8), (9)PC) and DSPE-PEG(2000) was developed for DXR encapsulation. Almost 70% of the small molecule therapeutic was retained. DXR was only released after a 514 nm laser treatment, in a wavelength specific manner. Furthermore, co-cultures containing DXR loaded liposomes with cells (Raji and MCF-7) showed improved cellular deaths after laser treatment, of at least 2 to 3 times higher compared to their untreated counterparts. Development of such nano-carriers with active release mechanism showed potential in reduced toxicity and thus could result in more effective treatments in the future.100
Gold-coated (Au) liposomes are another appealing method for light triggered release of DXR. This is due to Au nanoparticles are able to generate heat upon exposure to near-infrared light irradiation. However, Au nanoshells with solid silica cores alone are unable to carry sufficient volumes of small molecule therapeutics. Therefore, liposome, prepared with soya phosphatidylcholine (SPC) and Chol, after being covered by silica, is further coated with Au as delivery carrier. The DXR release was improved via illumination of a 808 nm laser beam (100 mW) over a time span of 15 min. DOX-loaded Au liposomes exhibited 82% tumour cell growth inhibition, whereas the empty Au liposomes exhibited only 58% inhibition rates.222 In another example, a liposome formulation bearing both light and heat sensitive properties was developed. This formulation consisted of DXR together with hollow gold nanospheres and the liposomes. It demonstrated rapid release of the DXR small molecule therapeutic after light irradiation. Furthermore, higher cytotoxicity was detected after near infrared light laser irradiation in the treatment of tumour cells compared to the control groups, indicating higher rates of antitumor efficiency. This results may be due to possible phase transitions and increased permeability of the entire liposomal membrane due to heat diffusion activities of the hollow Au nanospheres on the liposomal membrane (Fig. 8).223
 | ||
| Fig. 8 Illustration for preparation of DOX-EL.209 | ||
4.4. Increasing liposome viability under extreme pH
While previously mentioned pHSL, liposomes are to target regions that has lower pH than normal physiological pH of 7.4, they may only exist stably at pH around 5.0.88,133 Some parts of the human body such as stomach's antral regions exhibit extremely low pH (as low as 1.2) due to the secretion of gastric fluids.226 At such extreme pH, conventional liposomes (including pHSL) will undergo decomposition and drug delivery will be compromised. Therefore, some studies have been done to increase liposome viability under such extreme pH. An efficient way to increase the liposome viability at low pH using polyelectrolyte multi-layered coatings is reported.227 In this study, the cationic liposomes were coated with poly(acrylic acid) (PAA) and poly(allylamine hydrochloride) (PAH) as polyelectrolyte and further used to encapsulate amoxicillin and metronidazole to treat helicobacter pylori infection at stomach antral. It showed that the encapsulation efficiency remained high for both formulations. Under simulated gastric fluid, the polyelectrolyte coated liposomes were able to remain viable at pH 1.2 and release drugs to inhibit in vivo bacterial growth up to 99%. In another study, a colon-specific liposomes coated in calcium alginate gel beads was developed and used to encapsulate bee venom to be taken orally.228 While alginate is a biodegradable compound that is safe for consumption, it showed that liposomes coated in the beads remained viable throughout the digestive system (including stomach), and successfully released at colon where pH is higher (pH 6.8–7.4). Fig. 9 shows the release curves of bee venom changes at different pH. As shown in the figure, the release is negligible at gastric pH of 1.2, and release begins at higher pH above 6.8.228 | ||
| Fig. 9 Graph on release of bee venom against pH and time.228 | ||
4.5. Clinical trials and commercial potential
Understanding that liposome is highly flexible in terms of compositional modification and surface engineering, it is considered as one of the most useful and most extensively studied amongst different kinds of nanocarriers. With Doxil® (PEGylated liposomal doxorubicin) as one example, liposome is able to carry small molecule therapeutics effectively, and target the desired cells with less chance of in vivo degradation and exhibits reduced chances of side effects, which makes it favourable as nanocarriers for many different small molecule therapeutics. However, while Doxil® has made astonishing results in the treatment for Acquired Immune Deficiency Syndrome (AIDS) related diseases and cancers in terms of delivery efficiency, the overall alleviating effect for the disease remains approximately the same in vivo condition. The reason being is the slow release property from PEGylation that results in reduced bioavailability of the small molecule therapeutic.199 Such limitations were minimized for another small molecule therapeutic called Myocet®, which is a type of DOX-encapsulated liposome without addition of PEG. This allowed rapid release with increased encapsulation of up to 95%. Furthermore, Lipo-Dox®, a similar variant of Doxil®, was formulated based on DSPC which has very close phase transition temperature to Doxil®,224 but due to compositional difference, there is some variation in the pharmacokinetics. In comparison to free small molecule therapeutic which exhibits half-life of about 0.2 hour, Myocet® has 2.5 hours, 55 hours for Doxil® and 65 h for LipoDox® respectively.33,224,225 Apart from these small molecule therapeutics, many others have successfully loaded into liposome in order to target cancerous cells, as shown in Table 1.25,33| Products | Route of injection | Small molecule therapeutics | Lipid composition | Indicators | Ref. |
|---|---|---|---|---|---|
| Exparel | Infiltration | Bupivacaine HCl | DPPG, tricaprylin, DEPC and Chol | Postsurgical analgesia | 227 |
| Depocyts | Spinal | Cytarabine | DPOC, Chol, triolein, DPPG | Lymphomatous meningitis, neoplastic meningitis | 14, 15, 25 and 33 |
| DaunoXome | Intravenous | Dauorubicin | DSPC and Chol | Advanced HIV-associated Kaposi sarcoma | 14 and 15 |
| Myocet | Intravenous | Doxorubicin | EPC and Chol | Metastatic breast cancer | 25 and 33 |
| Doxil/Caelyx | Intravenous | Doxorubicin | HSPC, Chol, PEG 2000-DSPE | Kaposi's sarcoma | 14, 15, 25 and 33 |
| Lipo-dox | Intravenous | Doxorubicin | DSPC, Chol, PEG 2000-DSPE | Ovarian cancer and AIDS related-Kaposi sarcoma | 14, 15, 25, 33 and 34 |
| Depodur | Epidural | Morphine sulphate | DOPC, Chol, DPPG and triolein | Pain following major surgery | 25 and 33 |
| Marqibo | Intravenous | Vincristine sulphate | Sphingomyelin and Chol | Philadelphia chromosome-negative (Ph-) acute lymphoblastic leukaemia | 14, 15, 25 and 226 |
One of the latest small molecule therapeutic-encapsulating liposome approved by FDA in August 2012 is Marqibo®, which encapsulated vincristine sulphate to treat patients with Philadelphia chromosome-negative acute lymphoblastic leukaemia.25,226 Furthermore, there are many other clinical trials for different liposomal products to alleviate various chronic diseases like cancer, cardiac and pulmonary illnesses. One example is ThermoDox®, which has function of heat-triggered release and is formulated from DPPC/MPPC/DSPE-PEG-2000. The trial results revealed that triggered release was indeed necessary and effective strategy to optimise the penetration and release of DOX-encapsulated liposomes.199 Even though ThermoDox® is still at its therapeutic testing for a few cancer treatments such as colorectal liver metastatic cancer (Phase II), breast cancer (Phase II) and primary liver cancer (Phase III), researches have shown the safety and efficacy of ThermoDox® in patients with aid of radio frequency ablation (RFA).
Other than ThermoDox®, there are other liposomal small molecule therapeutics undergoing clinical trials such as alendronate encapsulated liposome to cure coronary artery steonosis which is at Phase II of the test and many others at Phase I of trial. Irinotecan liposomes (nanoliposomal CPT-11) were developed based on remote loading but with a modified gradient comprising with a polyalkylammonium salt along with a polymeric (poly-phosphate) or nonpolymeric (sucrose octasulphate) anion as intraliposomal trapping agents. The formulation is able to maintain the small molecule therapeutic encapsulated when administered in vivo with a half-life of 56.8 h.227 The clinical trials with nanoliposomal CPT-11 are currently in Phase I for treatment of glioblastoma, gliosarcoma and anaplastic astrocytoma among other types of cancer.221 PEGylated irinotecan (IHL-305) for the treatment of solid tumours222 and topotecan liposomes for the treatment of small cell lung cancer, ovarian cancer and solid tumours are also under Phase I clinical trials.223
Liposomes loaded with specifically irinotecan small molecule therapeutics (nanoliposomal CPT-11) were further progressed with reference to the mechanisms of remote loading, however, with a different gradient, where in this case, comprises of a polyalkyl ammonium salt together with either a polymeric (poly-phosphate) or with a non-polymeric (sucrose octasulphate) anion as trapping agents within the liposome. This composition of liposome was found to stabilise the encapsulation of the small molecule therapeutics during in vivo delivery, bearing a half-life of 56.8 hours.227 Nanoliposomal CPT-11 is presently undergoing clinical tests specifically catered to treat several types of cancer, such as glioblastoma, gliosarcoma and anaplastic astrocytoma.223 In addition, small cell lung cancer, ovarian cancer, solid tumours are also undergoing Phase I of clinical tests with the use of topotecan liposomes,224 while PEGylated irinotecan (IHL-305) is undergoing tests as a treatment method for solid tumours.223
5. Conclusion and future perspectives
For this review, two general categories regarding the small molecule therapeutics loading and the methods to encapsulate and to deliver small molecule therapeutics to improve their therapeutic functions were discussed. Further categorising into two subgroups, small molecule therapeutics encapsulation efficiencies can be affected either via passive means or active approaches. While variety of techniques are available to achieve as such encapsulation and delivery path, each techniques shares different pros and cons and opposing effects. In case of increasing encapsulation efficiency, increasing stability of liposomes usually tends to increase leading to decrease in release potential. To minimize this limitation, different effective triggered release methods were also discussed. This review also stated various hydrophilic small molecule therapeutics that have successfully encapsulated in liposome and their respective experimental effects in vitro and in vivo. Some of them, such as Doxil® are already clinically available while many more are under trials. Although they are still under trial, their experimental results proves that liposomal small molecule therapeutics have exceptional potential as therapeutic medicine. In conclusion, liposomal encapsulation provides various ways of modification, which provides efficient encapsulation that increases half-life of small molecule therapeutics, target delivery and release, and lastly increases effective expression of the small molecule therapeutics. These liposomes have to contend with the likes of micelle228–234 and gel-based delivery agents235–242 and further development has to be carried out to carve out a niche area for liposome delivery therapies.Previous studies have shown the great success of using these anti-cancer drugs loaded liposomes to achieve enhanced therapeutic effects and reduced side effects compared to more traditional dosage forms. Although the drug loading or encapsulating efficiency in liposomal drug development have been significantly improved, some of the remaining critical challenges should be addressed before it can moves to the next stage on the development of liposome based drug delivery system for wide adoption. As a typical concern highlighted here, the liposome-specific adverse effects such as the commonly occurred skin reactions and hypersensitivity reactions must be considered. For example, skin toxicity and hand-foot syndrome were observed after the injection of doxorubicin loaded PEG-liposomes, indicating the effects of DOX were hindered due to the sustained circulation and high stability of the liposome.243 Therefore, it is suggested the long circulating liposomes may cause the accumulation mainly in the reigns of palms, soles and areas of repeated friction or trauma, which would further mediate the release of DOX from liposomes under these conditions.244 In addition, despite the preventive measures in the first cycle of chemotherapy, acute hypersensitivity associated with infusions of liposomal DOX was also detected in an ovarian cancer patient. Therefore, it should be mandatory to closely monitor the signs of hypersensitivity during the first 15 min for patients who receive their first dose of liposomal DOX.245 Similar studies also showed 30.8% of patients had hypersensitivity reactions including cutaneous reactions, respiratory, hemodynamic, hypotension etc. These types of reactions are likely due to the activation of complement immunity.246 At present, it is required to understand the fundamentals about the induction of liposome-specific adverse effects and to develop proper methods to alleviate these unwanted side effects. Further research should also identify patients who are at risk of experiencing such hypersensitivity reactions.247
As for the targeting delivery, in addition to the accumulation of drugs-loaded liposomes in the interstitial space inside tumours, the ligand-mediated tumour targeting liposomes have to be effectively internalized by the target cells to further allow the deposition of drugs at high intracellular concentration and bypassing multidrug resistance. To achieve this aim, there are certain requirements need to be considered for the future development of new ligand-mediated tumour targeting liposomes. For example, the target should be identified in sufficient quantity and overexpressed on the tumor cells surface, which can provide high binding affinity between the liposomes and targeted cancer cells. In addition, the attachment of specific ligand to the surface of the drug-loaded liposomes should not alter the binding affinity as well as the long circulation property of liposome in blood. Moreover, the targeting ligand should be internalizable and the presence of ligand could assist the cell internalization of the liposomes and the anti-cancer loaded liposomes. Finally, upon up-taken by cancer cells, the drugs released from the liposomes should maintain above the therapeutic concentration and sustain the release for a predesigned period of time to achieve efficient cancer therapy. With further studies and optimizations of these design parameters, we could see the dawn of the wide applications of liposomes as delivery carriers to cure diseases around the world in the future.
References
- X. Xu, M. A. Khan and D. J. Burgess, Int. J. Pharm., 2012, 423, 543–553 CrossRef CAS PubMed
.
- S. Vrignaud, J.-P. Benoit and P. Saulnier, Biomaterials, 2011, 32, 8593–8604 CrossRef CAS PubMed
- Z. Li, D. Yuan, G. Jin, B. H. Tan and C. He, ACS Appl. Mater. Interfaces, 2016, 8, 1842–1853 CAS
- Z. Li, J. Yang and X. J. Loh, NPG Asia Mater., 2016, 8, e265 CrossRef CAS
- Z. Li, B. H. Tan, T. Lin and C. He, Prog. Polym. Sci., 2016 DOI:10.1016/j.progpolymsci.2016.05.003
- Z. Li and L. W. Swee, Mater. Sci. Eng., C, 2016 DOI:10.1016/j.msec.2016.03.039
- Z. Li and L. Wu, in Polyhydroxyalkanoates (PHAs): Biosynthesis, Industrial Production and Applications in Medicine, ed. W. Linping, Nova Science Publishers, USA, 2014, ch. 17, pp. 181–217 Search PubMed
- Z. Li and J. Li, J. Phys. Chem. B, 2013, 117, 14763–14774 CrossRef CAS PubMed
- A. Patel and K. P. Velikov, LWT--Food Sci. Technol., 2011, 44, 1958–1964 CrossRef CAS
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS PubMed
- A. Johansson, J. Svensson, N. Bendsoe, K. Svanberg, E. Alexandratou, M. Kyriazi, D. Yova, S. Grafe, T. Trebst and S. Andersson-Engels, J. Biomed. Opt., 2007, 12, 034026 CrossRef PubMed
- T. Yamamoto, M. Yokoyama, P. Opanasopit, A. Hayama, K. Kawano and Y. Maitani, J. Controlled Release, 2007, 123, 11–18 CrossRef CAS PubMed
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS
- A. L. Klibanov, K. Maruyama, V. P. Torchilin and L. Huang, FEBS Lett., 1990, 268, 235–237 CrossRef CAS PubMed
- T. Allen, C. Hansen, F. Martin, C. Redemann and A. Yau-Young, Biochim. Biophys. Acta, Biomembr., 1991, 1066, 29–36 CrossRef CAS
- Z. Li, P. L. Chee, C. Owh, R. Lakshminarayanan and X. J. Loh, RSC Adv., 2016, 6, 28947–28955 RSC
- X. Su, M. J. Tan, Z. Li, M. Wong, L. Rajamani, G. Lingam and X. J. Loh, Biomacromolecules, 2015, 16, 3093–3102 CrossRef CAS PubMed
- X. Fan, Z. Wang, D. Yuan, Y. Sun, Z. Li and C. He, Polym. Chem., 2014, 5, 4069–4075 RSC
- Z. Li, H. Yin, Z. Zhang, K. L. Liu and J. Li, Biomacromolecules, 2012, 13, 3162–3172 CrossRef CAS PubMed
- L. Mayer, M. Hope, P. Cullis and A. Janoff, Biochim. Biophys. Acta, Biomembr., 1985, 817, 193–196 CrossRef CAS
- R. Cortesi, J. Microencapsulation, 1999, 16, 251–256 CrossRef CAS PubMed
- Y. C. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed
- G. Haran, R. Cohen, L. K. Bar and Y. Barenholz, Biochim. Biophys. Acta, Biomembr., 1993, 1151, 201–215 CrossRef CAS
- S. Bibi, E. Lattmann, A. R. Mohammed and Y. Perrie, J. Microencapsulation, 2012, 29, 262–276 CrossRef CAS PubMed
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed
- M. Kępczyński, K. Nawalany, M. Kumorek, A. Kobierska, B. Jachimska and M. Nowakowska, Chem. Phys. Lipids, 2008, 155, 7–15 CrossRef PubMed
- M. Glavas-Dodov, E. Fredro-Kumbaradzi, K. Goracinova, M. Simonoska, S. Calis, S. Trajkovic-Jolevska and A. Hincal, Int. J. Pharm., 2005, 291, 79–86 CrossRef CAS PubMed
- V. Manojlovic, K. Winkler, V. Bunjes, A. Neub, R. Schubert, B. Bugarski and G. Leneweit, Colloids Surf., B, 2008, 64, 284–296 CrossRef CAS PubMed
- K. Taylor, G. Taylor, I. Kellaway and J. Stevens, Int. J. Pharm., 1990, 58, 49–55 CrossRef CAS
- A. Di Giulio, G. Maurizi, P. Odoardi, M. Saletti, G. Amicosante and A. Oratore, Int. J. Pharm., 1991, 74, 183–188 CrossRef CAS
- W. C. Zamboni, A. C. Gervais, M. J. Egorin, J. H. Schellens, E. G. Zuhowski, D. Pluim, E. Joseph, D. R. Hamburger, P. K. Working and G. Colbern, Cancer Chemother. Pharmacol., 2004, 53, 329–336 CrossRef CAS PubMed
- A. Chaudhury, S. Das, R. F. Lee, K.-B. Tan, W.-K. Ng, R. B. Tan and G. N. Chiu, Int. J. Pharm., 2012, 430, 167–175 CrossRef CAS PubMed
- H.-I. Chang and M.-K. Yeh, Int. J. Nanomed., 2012, 7, 49–60 CAS
- K.-H. Chuang, H.-E. Wang, F.-M. Chen, S.-C. Tzou, C.-M. Cheng, Y.-C. Chang, W.-L. Tseng, J. Shiea, S.-R. Lin and J.-Y. Wang, Mol. Cancer Ther., 2010, 9, 1903–1912 CrossRef CAS PubMed
- A. Gürsoy, E. Kut and S. Özkırımlı, Int. J. Pharm., 2004, 271, 115–123 CrossRef
- H.-K. Han, H.-J. Shin and D. H. Ha, Eur. J. Pharm. Sci., 2012, 46, 500–507 CrossRef CAS PubMed
- B. Moghaddam, M. H. Ali, J. Wilkhu, D. J. Kirby, A. R. Mohammed, Q. Zheng and Y. Perrie, Int. J. Pharm., 2011, 417, 235–244 CrossRef CAS PubMed
- T. Schneider, A. Sachse, G. Röβling and M. Brandl, Int. J. Pharm., 1995, 117, 1–12 CrossRef CAS
- Y. Wang, W. Su, Q. Li, C. Li, H. Wang, Y. Li, Y. Cao, J. Chang and L. Zhang, Int. J. Pharm., 2013, 441, 748–756 CrossRef CAS PubMed
- S. Villasmil-Sánchez, A. M. Rabasco and M. L. González-Rodríguez, Colloids Surf., B, 2013, 105, 14–23 CrossRef PubMed
- E. Kajiwara, K. Kawano, Y. Hattori, M. Fukushima, K. Hayashi and Y. Maitani, J. Controlled Release, 2007, 120, 104–110 CrossRef CAS PubMed
- B. Ruozi, G. Riva, D. Belletti, G. Tosi, F. Forni, A. Mucci, P. Barozzi, M. Luppi and M. Vandelli, Eur. J. Pharm. Sci., 2010, 41, 254–264 CrossRef CAS PubMed
- A. Bangham, M. M. Standish and J. Watkins, J. Mol. Biol., 1965, 13, 238–252 CrossRef CAS PubMed
- F. Szoka and D. Papahadjopoulos, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 4194–4198 CrossRef CAS
- X. Xu, M. A. Khan and D. J. Burgess, Int. J. Pharm., 2012, 423, 410–418 CrossRef CAS PubMed
- B. Zadi and G. Gregoriadis, J. Liposome Res., 2000, 10, 73–80 CrossRef CAS
- J. D. Castile and K. M. Taylor, Int. J. Pharm., 1999, 188, 87–95 CrossRef CAS PubMed
- C. J. Chapman, W. L. Erdahl, R. W. Taylor and D. R. Pfeiffer, Chem. Phys. Lipids, 1990, 55, 73–83 CrossRef CAS PubMed
- K. Anzai, M. Yoshida and Y. Kirino, Biochim. Biophys. Acta, Biomembr., 1990, 1021, 21–26 CrossRef CAS
- J. O. Eloy, M. C. de Souza, R. Petrilli, J. P. A. Barcellos, R. J. Lee and J. M. Marchetti, Colloids Surf., B, 2014, 123, 345–363 CrossRef CAS PubMed
- M. Ueno and S. Sriwongsitanont, Polymer, 2005, 46, 1257–1267 CrossRef CAS
- H. Agashe, P. Lagisetty, S. Awasthi and V. Awasthi, Colloids Surf., B, 2010, 75, 573–583 CrossRef CAS PubMed
- M. Hope, M. Bally, G. Webb and P. Cullis, Biochim. Biophys. Acta, Biomembr., 1985, 812, 55–65 CrossRef CAS
- A. Akbarzadeh, R. Rezaei-Sadabady, S. Davaran, S. W. Joo, N. Zarghami, Y. Hanifehpour, M. Samiei, M. Kouhi and K. Nejati-Koshki, Nanoscale Res. Lett., 2013, 8, 102 CrossRef PubMed
- S. Vemuri and C. Rhodes, Pharm. Acta Helv., 1995, 70, 95–111 CrossRef CAS PubMed
- B. Elorza, M. Elorza, G. Frutos and J. Chantres, Biochim. Biophys. Acta, Biomembr., 1993, 1153, 135–142 CrossRef CAS
- L. Zhang, L. Han, X. Sun, D. Gao, J. Qin and J. Wang, Fitoterapia, 2012, 83, 678–689 CrossRef CAS PubMed
- K. Muppidi, A. S. Pumerantz, J. Wang and G. Betageri, ISRN Pharm., 2012, 2012 Search PubMed
- Y. P. Patil and S. Jadhav, Chem. Phys. Lipids, 2014, 177, 8–18 CrossRef CAS PubMed
- R. R. Hood, C. Shao, D. M. Omiatek, W. N. Vreeland and D. L. DeVoe, Pharm. Res., 2013, 30, 1597–1607 CrossRef CAS PubMed
- G. Yang, Y. Zhao, Y. Zhang, B. Dang, Y. Liu and N. Feng, Int. J. Nanomed., 2015, 10, 6633–6644 CrossRef CAS PubMed
- I. E. Santo, R. Campardelli, E. C. Albuquerque, S. A. Vieira De Melo, E. Reverchon and G. Della Porta, J. Pharm. Sci., 2015, 104, 3842–3850 CrossRef CAS PubMed
- F. Tao and P. Yang, Sci. Rep., 2015, 5, 9839 CrossRef CAS PubMed
- E. M. Schmid, D. L. Richmond and D. A. Fletcher, Methods Cell Biol., 2015, 128, 319–338 Search PubMed
- K. Karamdad, R. V. Law, J. M. Seddon, N. J. Brooks and O. Ces, Lab Chip, 2015, 15, 557–562 RSC
- M. Breton, M. Amirkavei and L. M. Mir, J. Membr. Biol., 2015, 248, 827–835 CrossRef CAS PubMed
- T. Wang, Y. Deng, Y. Geng, Z. Gao, J. Zou and Z. Wang, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 222–231 CrossRef CAS PubMed
- T. Kuroiwa, H. Kiuchi, K. Noda, I. Kobayashi, M. Nakajima, K. Uemura, S. Sato, S. Mukataka and S. Ichikawa, Microfluid. Nanofluid., 2009, 6, 811–821 CrossRef CAS
- R. k. Genç, M. Ortiz and C. K. O'Sullivan, Langmuir, 2009, 25, 12604–12613 CrossRef PubMed
- H. C. Shum, D. Lee, I. Yoon, T. Kodger and D. A. Weitz, Langmuir, 2008, 24, 7651–7653 CrossRef CAS PubMed
- R. T. Davies, D. Kim and J. Park, J. Micromech. Microeng., 2012, 22, 055003 CrossRef
- J. W. Nichols and D. W. Deamer, Biochim. Biophys. Acta, Biomembr., 1976, 455, 269–271 CrossRef CAS
- D. W. Deamer, R. C. Prince and A. R. Crofts, Biochim. Biophys. Acta, Biomembr., 1972, 274, 323–335 CrossRef CAS
- S. Hwang, Y. Maitani, X.-R. Qi, K. Takayama and T. Nagai, Int. J. Pharm., 1999, 179, 85–95 CrossRef CAS PubMed
- D. Zucker, D. Marcus, Y. Barenholz and A. Goldblum, J. Controlled Release, 2009, 139, 73–80 CrossRef CAS PubMed
- X. Li, D. J. Hirsh, D. Cabral-Lilly, A. Zirkel, S. M. Gruner, A. S. Janoff and W. R. Perkins, Biochim. Biophys. Acta, Biomembr., 1998, 1415, 23–40 CrossRef CAS
- S. Clerc and Y. Barenholz, Anal. Biochem., 1998, 259, 104–111 CrossRef CAS PubMed
- A. Fritze, F. Hens, A. Kimpfler, R. Schubert and R. Peschka-Süss, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1633–1640 CrossRef CAS PubMed
- S. Clerc and Y. Barenholz, Biochim. Biophys. Acta, Biomembr., 1995, 1240, 257–265 CrossRef
- S. A. Abraham, K. Edwards, G. Karlsson, N. Hudon, L. D. Mayer and M. B. Bally, J. Controlled Release, 2004, 96, 449–461 CrossRef CAS PubMed
- C. Li, J. Cui, Y. Li, C. Wang, Y. Li, L. Zhang, L. Zhang, W. Guo, J. Wang and H. Zhang, Eur. J. Pharm. Sci., 2008, 34, 333–344 CrossRef CAS PubMed
- J. Cui, C. Li, L. Wang, C. Wang, H. Yang, Y. Li, L. Zhang, L. Zhang, W. Guo and M. Liang, Int. J. Pharm., 2009, 368, 24–30 CrossRef CAS PubMed
- A. Cern, A. Golbraikh, A. Sedykh, A. Tropsha, Y. Barenholz and A. Goldblum, J. Controlled Release, 2012, 160, 147–157 CrossRef CAS PubMed
- H. Maeda, J. Fang, T. Inutsuka and Y. Kitamoto, Int. Immunopharmacol., 2003, 3, 319–328 CrossRef CAS PubMed
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed
- T. N. Palmer, V. J. Caride, M. A. Caldecourt, J. Twickler and V. Abdullah, Biochim. Biophys. Acta, Gen. Subj., 1984, 797, 363–368 CrossRef CAS
- C. Kelly, C. Jefferies and S.-A. Cryan, J. Drug Delivery, 2010, 2011 Search PubMed
- T. L. Andresen, S. S. Jensen and K. Jørgensen, Prog. Lipid Res., 2005, 44, 68–97 CrossRef CAS PubMed
- H. Zhang, Z.-y. Wang, W. Gong, Z.-p. Li, X.-g. Mei and W.-l. Lv, Int. J. Pharm., 2011, 414, 56–62 CrossRef CAS PubMed
- Y. Wu, Y. Yang, F.-c. Zhang, C. Wu, W.-L. Lü and X.-G. Mei, J. Liposome Res., 2011, 21, 221–228 CrossRef CAS PubMed
- L. Li, T. L. ten Hagen, M. Hossann, R. Süss, G. C. van Rhoon, A. M. Eggermont, D. Haemmerich and G. A. Koning, J. Controlled Release, 2013, 168, 142–150 CrossRef CAS PubMed
- E. Torres, F. Mainini, R. Napolitano, F. Fedeli, R. Cavalli, S. Aime and E. Terreno, J. Controlled Release, 2011, 154, 196–202 CrossRef CAS PubMed
- M. J. Morilla, J. Montanari, F. Frank, E. Malchiodi, R. Corral, P. Petray and E. L. Romero, J. Controlled Release, 2005, 103, 599–607 CrossRef CAS PubMed
- S. M. Z. M. D. Ferreira, G. P. Domingos, D. dos Santos Ferreira, T. G. R. Rocha, R. Serakides, C. M. de Faria Rezende, V. N. Cardoso, S. O. A. Fernandes and M. C. Oliveira, Bioorg. Med. Chem. Lett., 2012, 22, 4605–4608 CrossRef CAS PubMed
- P. H. Kee, T. A. Abruzzo, D. A. Smith, J. A. Kopechek, B. Wang, S. L. Huang, R. C. MacDonald, C. K. Holland and D. D. McPherson, J. Liposome Res., 2008, 18, 263–277 CrossRef CAS PubMed
- S.-L. Huang, D. D. McPherson and R. C. MacDonald, Ultrasound Med. Biol., 2008, 34, 1272–1280 CrossRef PubMed
- S.-L. Huang and R. C. MacDonald, Biochim. Biophys. Acta, Biomembr., 2004, 1665, 134–141 CrossRef CAS PubMed
- S. Nappini, F. B. Bombelli, M. Bonini, B. Nordèn and P. Baglioni, Soft Matter, 2010, 6, 154–162 RSC
- M. Babincova, P. Čičmanec, V. Altanerova, Č. Altaner and P. Babinec, Bioelectrochemistry, 2002, 55, 17–19 CrossRef CAS PubMed
- A. Yavlovich, A. Singh, R. Blumenthal and A. Puri, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 117–126 CrossRef CAS PubMed
- F. Yan, L. Li, Z. Deng, Q. Jin, J. Chen, W. Yang, C.-K. Yeh, J. Wu, R. Shandas and X. Liu, J. Controlled Release, 2013, 166, 246–255 CrossRef CAS PubMed
- P. Kulshrestha, M. Gogoi, D. Bahadur and R. Banerjee, Colloids Surf., B, 2012, 96, 1–7 CrossRef CAS PubMed
- J. A. Kopechek, T. M. Abruzzo, B. Wang, S. M. Chrzanowski, D. A. Smith, P. H. Kee, S. Huang, J. H. Collier, D. D. McPherson and C. K. Holland, J. Ultrasound Med., 2008, 27, 1597–1606 Search PubMed
- D. C. Drummond, O. Meyer, K. Hong, D. B. Kirpotin and D. Papahadjopoulos, Pharmacol. Rev., 1999, 51, 691–744 CAS
- S. A. Durazo and U. B. Kompella, Mitochondrion, 2012, 12, 190–201 CrossRef CAS PubMed
- E. Koren, A. Apte, A. Jani and V. P. Torchilin, J. Controlled Release, 2012, 160, 264–273 CrossRef CAS PubMed
- H. Maeda, L. W. Seymour and Y. Miyamoto, Bioconjugate Chem., 1992, 3, 351–362 CrossRef CAS PubMed
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed
- R. K. Jain, Annu. Rev. Biomed. Eng., 1999, 1, 241–263 CrossRef CAS PubMed
- T. Tanaka, P. Decuzzi, M. Cristofanilli, J. H. Sakamoto, E. Tasciotti, F. M. Robertson and M. Ferrari, Biomed. Microdevices, 2009, 11, 49–63 CrossRef CAS PubMed
- R. K. Jain, Adv. Drug Delivery Rev., 2001, 46, 149–168 CrossRef CAS PubMed
- S. Stapleton, M. Milosevic, C. Allen, J. Zheng, M. Dunne, I. Yeung and D. A. Jaffray, PLoS One, 2013, 8, e81157 Search PubMed
- K. Maruyama, Adv. Drug Delivery Rev., 2011, 63, 161–169 CrossRef CAS PubMed
- C. Li and S. Wallace, Adv. Drug Delivery Rev., 2008, 60, 886–898 CrossRef CAS PubMed
- C. E. Petre and D. P. Dittmer, Int. J. Nanomed., 2007, 2, 277 CAS
- S. Biswas, N. S. Dodwadkar, P. P. Deshpande, S. Parab and V. P. Torchilin, Eur. J. Pharm. Biopharm., 2013, 84, 517–525 CrossRef CAS PubMed
- Z. Yan, F. Wang, Z. Wen, C. Zhan, L. Feng, Y. Liu, X. Wei, C. Xie and W. Lu, J. Controlled Release, 2012, 157, 118–125 CrossRef CAS PubMed
- M. Yokoyama, J. Exp. Clin. Med., 2011, 3, 151–158 CrossRef CAS
- K. Shiraishi, K. Kawano, Y. Maitani and M. Yokoyama, J. Controlled Release, 2010, 148, 160–167 CrossRef CAS PubMed
- K. Shiraishi, K. Kawano, T. Minowa, Y. Maitani and M. Yokoyama, J. Controlled Release, 2009, 136, 14–20 CrossRef CAS PubMed
- E. Nakamura, K. Makino, T. Okano, T. Yamamoto and M. Yokoyama, J. Controlled Release, 2006, 114, 325–333 CrossRef CAS PubMed
- L. Holmgren, M. S. O'Reilly and J. Folkman, Nat. Med., 1995, 1, 149–153 CrossRef CAS PubMed
- J. Folkman, in Biology of endothelial cells, Springer, 1984, pp. 412–428 Search PubMed
- N. Monteiro, A. Martins, R. L. Reis and N. M. Neves, J. R. Soc., Interface, 2014, 11, 20140459 CrossRef
- J. A. Harding, C. M. Engbers, M. S. Newman, N. I. Goldstein and S. Zalipsky, Biochim. Biophys. Acta, Biomembr., 1997, 1327, 181–192 CrossRef CAS
- G. A. Koning, J. A. Kamps and G. L. Scherphof, J. Liposome Res., 2002, 12, 107–119 CrossRef CAS PubMed
- E. Vives, P. Brodin and B. Lebleu, J. Biol. Chem., 1997, 272, 16010–16017 CrossRef CAS PubMed
- H. Nagahara, A. M. Vocero-Akbani, E. L. Snyder, A. Ho, D. G. Latham, N. A. Lissy, M. Becker-Hapak, S. A. Ezhevsky and S. F. Dowdy, Nat. Med., 1998, 4, 1449–1452 CrossRef CAS PubMed
- W. C. Chen, D. S. Sigal, A. Saven and J. C. Paulson, Leuk. Lymphoma, 2012, 53, 208–210 CrossRef CAS PubMed
- D. Peer and R. Margalit, Int. J. Cancer, 2004, 108, 780–789 CrossRef CAS PubMed
- X. Q. Pan, H. Wang and R. J. Lee, Pharm. Res., 2003, 20, 417–422 CrossRef CAS
- K. Watanabe, M. Kaneko and Y. Maitani, Int. J. Nanomed., 2012, 7, 3679 CAS
- D. C. Drummond, M. Zignani and J.-C. Leroux, Prog. Lipid Res., 2000, 39, 409–460 CrossRef CAS PubMed
- A. A. Karim, Q. Dou, Z. Li and X. J. Loh, Chem.–Asian J., 2016 DOI:10.1002/asia.201501434
- Z. Li, D. Yuan, X. Fan, B. H. Tan and C. He, Langmuir, 2015, 31, 2321–2333 CrossRef CAS PubMed
- Z. Li and X. J. Loh, Chem. Soc. Rev., 2015, 44, 2865–2879 RSC
- F. Perche and V. P. Torchilin, J. Drug Delivery, 2013, 2013 Search PubMed
- P. P. Deshpande, S. Biswas and V. P. Torchilin, Nanomedicine, 2013, 8, 1509–1528 CrossRef CAS PubMed
- S. Simões, V. Slepushkin, N. Düzgünes and M. C. P. de Lima, Biochim. Biophys. Acta, Biomembr., 2001, 1515, 23–37 CrossRef
- D. C. Litzinger and L. Huang, Biochim. Biophys. Acta, Rev. Biomembr., 1992, 1113, 201–227 CrossRef CAS
- E. Ducat, J. Deprez, A. Gillet, A. Noël, B. Evrard, O. Peulen and G. Piel, Int. J. Pharm., 2011, 420, 319–332 CrossRef CAS PubMed
- E. A. Kotova, A. V. Kuzevanov, A. A. Pashkovskaya and Y. N. Antonenko, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2252–2257 CrossRef CAS PubMed
- Y. Obata, S. Tajima and S. Takeoka, J. Controlled Release, 2010, 142, 267–276 CrossRef CAS PubMed
- A. M. Gomez, Fusogenic anti-PSMA liposomes for antivascular chemotherapy, Rutgers University-Graduate School-New Brunswick, 2013 Search PubMed
- A. L. B. de Barros, L. das Graças Mota, D. C. F. Soares, M. M. A. Coelho, M. C. Oliveira and V. N. Cardoso, Bioorg. Med. Chem. Lett., 2011, 21, 7373–7375 CrossRef PubMed
- V. A. Carmo, C. S. Ferrari, E. C. Reis, G. A. Ramaldes, M. A. Pereira, M. C. De Oliveira and V. N. Cardoso, Nucl. Med. Commun., 2008, 29, 33–38 CrossRef CAS PubMed
- S. Banerjee, K. Sen, T. K. Pal and S. K. Guha, Int. J. Pharm., 2012, 436, 786–797 CrossRef CAS PubMed
- I. F. Tannock and D. Rotin, Cancer Res., 1989, 49, 4373–4384 CAS
- R. van Sluis, Z. M. Bhujwalla, N. Raghunand, P. Ballesteros, J. Alvarez, S. Cerdán, J.-P. Galons and R. J. Gillies, Magn. Reson. Med., 1999, 41, 743–750 CrossRef CAS PubMed
- D. Wijesinghe, M. C. Arachchige, A. Lu, Y. K. Reshetnyak and O. A. Andreev, Sci. Rep., 2013, 3, 3560 Search PubMed
- Y. K. Reshetnyak, O. A. Andreev, M. Segala, V. S. Markin and D. M. Engelman, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15340–15345 CrossRef CAS PubMed
- O. A. Andreev, D. M. Engelman and Y. K. Reshetnyak, Chim. Oggi, 2009, 27, 34 Search PubMed
- L. Yao, J. Daniels, D. Wijesinghe, O. A. Andreev and Y. K. Reshetnyak, J. Controlled Release, 2013, 167, 228–237 CrossRef CAS PubMed
- Y. K. Reshetnyak, M. Segala, O. A. Andreev and D. M. Engelman, Biophys. J., 2007, 93, 2363–2372 CrossRef CAS PubMed
- J. F. Hunt, P. Rath, K. J. Rothschild and D. M. Engelman, Biochemistry, 1997, 36, 15177–15192 CrossRef CAS PubMed
- O. A. Andreev, A. D. Dupuy, M. Segala, S. Sandugu, D. A. Serra, C. O. Chichester, D. M. Engelman and Y. K. Reshetnyak, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7893–7898 CrossRef CAS PubMed
- S. Karve, A. Bandekar, M. R. Ali and S. Sofou, Biomaterials, 2010, 31, 4409–4416 CrossRef CAS PubMed
- I.-Y. Kim, Y.-S. Kang, D. S. Lee, H.-J. Park, E.-K. Choi, Y.-K. Oh, H.-J. Son and J.-S. Kim, J. Controlled Release, 2009, 140, 55–60 CrossRef CAS PubMed
- M. Zignani, D. C. Drummond, O. Meyer, K. Hong and J.-C. Leroux, Biochim. Biophys. Acta, Biomembr., 2000, 1463, 383–394 CrossRef CAS
- F. Pétriat, E. Roux, J. C. Leroux and S. Giasson, Langmuir, 2004, 20, 1393–1400 CrossRef
- O. Meyer, D. Papahadjopoulos and J.-C. Leroux, FEBS Lett., 1998, 421, 61–64 CrossRef CAS PubMed
- M. F. Francis, G. Dhara, F. M. Winnik and J.-C. Leroux, Biomacromolecules, 2001, 2, 741–749 CrossRef CAS PubMed
- P. Simard and J.-C. Leroux, Int. J. Pharm., 2009, 381, 86–96 CrossRef CAS PubMed
- Y. Liu, L.-L. Li, G.-B. Qi, X.-G. Chen and H. Wang, Biomaterials, 2014, 35, 3406–3415 CrossRef CAS PubMed
- D. C. F. Soares, M. C. de Oliveira, A. L. B. de Barros, V. N. Cardoso and G. A. Ramaldes, Eur. J. Pharm. Sci., 2011, 43, 290–296 CrossRef CAS PubMed
- D. C. F. Soares, V. N. Cardoso, A. L. B. de Barros, C. M. de Souza, G. D. Cassali, M. C. de Oliveira and G. A. Ramaldes, Eur. J. Pharm. Sci., 2012, 45, 58–64 CrossRef CAS PubMed
- Z. M. Saiyed, N. H. Gandhi and M. P. Nair, Int. J. Nanomed., 2010, 5, 157–166 CAS
- S. J. Soenen, S. F. De Meyer, T. Dresselaers, G. V. Velde, I. M. Pareyn, K. Braeckmans, M. De Cuyper, U. Himmelreich and K. I. Vanhoorelbeke, Biomaterials, 2011, 32, 4140–4150 CrossRef CAS PubMed
- D. Frascione, C. Diwoky, G. Almer, P. Opriessnig, C. Vonach, K. Gradauer, G. Leitinger, H. Mangge, R. Stollberger and R. Prassl, Int. J. Nanomed., 2012, 7, 2349 CAS
- M. Faria, M. Cruz, M. Gonçalves, A. Carvalho, G. Feio and M. Martins, Int. J. Pharm., 2013, 446, 183–190 CrossRef CAS PubMed
- N. Kawai, A. Ito, Y. Nakahara, M. Futakuchi, T. Shirai, H. Honda, T. Kobayashi and K. Kohri, Prostate, 2005, 64, 373–381 CrossRef CAS PubMed
- S. Hamaguchi, I. Tohnai, A. Ito, K. Mitsudo, T. Shigetomi, M. Ito, H. Honda, T. Kobayashi and M. Ueda, Cancer Sci., 2003, 94, 834–839 CrossRef CAS PubMed
- M.-S. Martina, J.-P. Fortin, C. Ménager, O. Clément, G. Barratt, C. Grabielle-Madelmont, F. Gazeau, V. Cabuil and S. Lesieur, J. Am. Chem. Soc., 2005, 127, 10676–10685 CrossRef CAS PubMed
- W. T. Al-Jamal and K. Kostarelos, Nanomedicine, 2007, 2(1), 85–98 CrossRef CAS PubMed
- C. Riviere, M.-S. Martina, Y. Tomita, C. Wilhelm, A. Tran Dinh, C. Menager, E. Pinard, S. Lesieur, F. Gazeau and J. Seylaz, Radiology, 2007, 244, 439–448 CrossRef PubMed
- S. J. Soenen, D. Vercauteren, K. Braeckmans, W. Noppe, S. De Smedt and M. De Cuyper, ChemBioChem, 2009, 10, 257–267 CrossRef CAS PubMed
- S. J. Soenen, M. Hodenius and M. De Cuyper, Nanomedicine, 2009, 4(2), 177–191 CrossRef CAS PubMed
- K. Tanaka, A. Ito, T. Kobayashi, T. Kawamura, S. Shimada, K. Matsumoto, T. Saida and H. Honda, J. Biosci. Bioeng., 2005, 100, 112–115 CrossRef CAS PubMed
- B. Clares, R. A. Biedma-Ortiz, E. Sáez-Fernández, J. C. Prados, C. Melguizo, L. Cabeza, R. Ortiz and J. L. Arias, Eur. J. Pharm. Biopharm., 2013, 85, 329–338 CrossRef CAS PubMed
- M. Bonini, D. Berti and P. Baglioni, Curr. Opin. Colloid Interface Sci., 2013, 18, 459–467 CrossRef CAS
- D. Qiu and X. An, Colloids Surf., B, 2013, 104, 326–329 CrossRef CAS PubMed
- K. Nahar, S. Absar, B. Patel and F. Ahsan, Int. J. Pharm., 2014, 464, 185–195 CrossRef CAS PubMed
- G. D. Bothun, A. Lelis, Y. Chen, K. Scully, L. E. Anderson and M. A. Stoner, Nanomedicine: Nanotechnology, Biology and Medicine, 2011, 7, 797–805 CrossRef CAS PubMed
- S. Jain, V. Mishra, P. Singh, P. Dubey, D. Saraf and S. Vyas, Int. J. Pharm., 2003, 261, 43–55 CrossRef CAS PubMed
- W. Li, B. Su, S. Meng, L. Ju, L. Yan, Y. Ding, Y. Song, W. Zhou, H. Li and L. Tang, Eur. J. Radiol., 2011, 80, 598–606 CrossRef PubMed
- P. Pradhan, J. Giri, F. Rieken, C. Koch, O. Mykhaylyk, M. Döblinger, R. Banerjee, D. Bahadur and C. Plank, J. Controlled Release, 2010, 142, 108–121 CrossRef CAS PubMed
- M. B. Yatvin, J. N. Weinstein, W. H. Dennis and R. Blumenthal, Science, 1978, 202, 1290–1293 CAS
- J. Weinstein, R. Magin, M. Yatvin and D. Zaharko, Science, 1979, 204, 188–191 CAS
- T. Tagami, J. P. May, M. J. Ernsting and S.-D. Li, J. Controlled Release, 2012, 161, 142–149 CrossRef CAS PubMed
- A. M. Ponce, B. L. Viglianti, D. Yu, P. S. Yarmolenko, C. R. Michelich, J. Woo, M. B. Bally and M. W. Dewhirst, J. Natl. Cancer Inst., 2007, 99, 53–63 CrossRef CAS PubMed
- D. Needham, G. Anyarambhatla, G. Kong and M. W. Dewhirst, Cancer Res., 2000, 60, 1197–1201 CAS
- G. Kong, G. Anyarambhatla, W. P. Petros, R. D. Braun, O. M. Colvin, D. Needham and M. W. Dewhirst, Cancer Res., 2000, 60, 6950–6957 CAS
- B. Hildebrandt, P. Wust, O. Ahlers, A. Dieing, G. Sreenivasa, T. Kerner, R. Felix and H. Riess, Crit. Rev. Oncol. Hematol., 2002, 43, 33–56 CrossRef PubMed
- G. Kong and M. Dewhirst, Int. J. Hyperthermia, 1999, 15, 345–370 CrossRef CAS PubMed
- W. Gong, Z. Wang, N. Liu, W. Lin, X. Wang, D. Xu, H. Liu, C. Zeng, X. Xie and X. Mei, Biol. Pharm. Bull., 2011, 34, 1058–1064 CAS
- J. K. Mills and D. Needham, Biochim. Biophys. Acta, Biomembr., 2005, 1716, 77–96 CrossRef CAS PubMed
- M. de Smet, S. Langereis, S. van den Bosch and H. Grüll, J. Controlled Release, 2010, 143, 120–127 CrossRef CAS PubMed
- L. Li, T. L. ten Hagen, A. Haeri, T. Soullié, C. Scholten, A. L. Seynhaeve, A. M. Eggermont and G. A. Koning, J. Controlled Release, 2014, 174, 202–208 CrossRef CAS PubMed
- T. Tagami, M. J. Ernsting and S.-D. Li, J. Controlled Release, 2011, 152, 303–309 CrossRef CAS PubMed
- T. Tagami, W. D. Foltz, M. J. Ernsting, C. M. Lee, I. F. Tannock, J. P. May and S.-D. Li, Biomaterials, 2011, 32, 6570–6578 CrossRef CAS PubMed
- M. S. Kim, D.-W. Lee, K. Park, S.-J. Park, E.-J. Choi, E. S. Park and H. R. Kim, Colloids Surf., B, 2014, 116, 17–25 CrossRef CAS PubMed
- Z. Li, B. H. Tan, G. Jin, K. Li and C. He, Polym. Chem., 2014, 5, 6740–6753 RSC
- Z. Li and B. H. Tan, Mater. Sci. Eng., C, 2014, 45, 620–634 CrossRef CAS PubMed
- Z. Li, Z. Zhang, K. L. Liu, X. Ni and J. Li, Biomacromolecules, 2012, 13, 3977–3989 CrossRef CAS PubMed
- L. Paasonen, B. Romberg, G. Storm, M. Yliperttula, A. Urtti and W. E. Hennink, Bioconjugate Chem., 2007, 18, 2131–2136 CrossRef CAS PubMed
- K. Kono, R. Nakai, K. Morimoto and T. Takagishi, Biochim. Biophys. Acta, Biomembr., 1999, 1416, 239–250 CrossRef CAS
- K. Kono, T. Ozawa, T. Yoshida, F. Ozaki, Y. Ishizaka, K. Maruyama, C. Kojima, A. Harada and S. Aoshima, Biomaterials, 2010, 31, 7096–7105 CrossRef CAS PubMed
- K. Kono, S. Nakashima, D. Kokuryo, I. Aoki, H. Shimomoto, S. Aoshima, K. Maruyama, E. Yuba, C. Kojima and A. Harada, Biomaterials, 2011, 32, 1387–1395 CrossRef CAS PubMed
- C.-Y. Lin, M. Javadi, D. M. Belnap, J. R. Barrow and W. G. Pitt, Nanomedicine: Nanotechnology, Biology and Medicine, 2014, 10, 67–76 CrossRef CAS PubMed
- S. L. Huang, A. J. Hamilton, A. Nagaraj, S. D. Tiukinhoy, M. E. Klegerman, D. D. Mcpherson and R. C. Macdonald, J. Pharm. Sci., 2001, 90, 1917–1926 CrossRef CAS PubMed
- H. Alkan-Onyuksel, S. M. Demos, G. M. Lanza, M. J. Vonesh, M. E. Klegerman, B. J. Kane, J. Kuszak and D. D. McPherson, J. Pharm. Sci., 1996, 85, 486–490 CrossRef CAS PubMed
- C. M. Newman, A. Lawrie, A. F. Brisken and D. C. Cumberland, Echocardiography, 2001, 18, 339–347 CAS
- K. D. Buchanan, S.-L. Huang, H. Kim, D. D. McPherson and R. C. MacDonald, J. Controlled Release, 2010, 141, 193–198 CrossRef CAS PubMed
- J. U. Menon, P. Jadeja, P. Tambe, K. Vu, B. Yuan and K. T. Nguyen, Theranostics, 2013, 3, 152–166 CrossRef CAS PubMed
- Q. Q. Dou, C. P. Teng, E. Y. Ye and X. J. Loh, Int. J. Nanomed., 2015, 10, 419–432 CrossRef CAS PubMed
- Q. Q. Dou, X. T. Fang, S. Jiang, P. L. Chee, T. C. Lee and X. J. Loh, RSC Adv., 2015, 5, 46817–46822 RSC
- L. Zhu and V. P. Torchilin, Integr. Biol., 2013, 5, 96–107 RSC
- L. Jiang, C. R. R. Gan, J. Gao and X. J. Loh, Small, 2016, 12, 3609–3644 CrossRef CAS PubMed
- S. A. de Visscher, S. Kaščáková, H. S. de Bruijn, A. v. d. van den Heuvel, A. Amelink, H. J. Sterenborg, D. J. Robinson, J. L. Roodenburg and M. J. Witjes, Lasers Surg. Med., 2011, 43, 528–536 CrossRef PubMed
- A. Yavlovich, B. Smith, K. Gupta, R. Blumenthal and A. Puri, Mol. Membr. Biol., 2010, 27, 364–381 CrossRef CAS PubMed
- S. J. Leung and M. Romanowski, Theranostics, 2012, 2, 1020–1036 CrossRef CAS PubMed
- C. Wu, C. Yu and M. Chu, Int. J. Nanomed., 2011, 6, 807 CAS
- J. You, P. Zhang, F. Hu, Y. Du, H. Yuan, J. Zhu, Z. Wang, J. Zhou and C. Li, Pharm. Res., 2014, 31, 554–565 CrossRef CAS PubMed
- Y. Fan and Q. Zhang, Asian J. Pharm. Sci., 2013, 8, 81–87 CrossRef CAS
- A. Gabizon, R. Catane, B. Uziely, B. Kaufman, T. Safra, R. Cohen, F. Martin, A. Huang and Y. Barenholz, Cancer Res., 1994, 54, 987–992 CAS
- J. A. Silverman and S. R. Deitcher, Cancer Chemother. Pharmacol., 2013, 71, 555–564 CrossRef CAS PubMed
- D. C. Drummond, C. O. Noble, Z. Guo, K. Hong, J. W. Park and D. B. Kirpotin, Cancer Res., 2006, 66, 3271–3277 CrossRef CAS PubMed
- Y.-L. Wu, H. Wang, Y.-K. Qiu and X. J. Loh, RSC Adv., 2016, 6, 44506–44513 RSC
- P. Janarthanan, A. K. Veeramachineni, C. Owh and X. J. Loh, ChemPlusChem, 2016, 81, 504–514 CrossRef
- X. J. Loh, S. J. Ong, Y. T. Tung and H. T. Choo, Polym. Chem., 2013, 4, 2564–2574 RSC
- X. J. Loh, S. J. Ong, Y. T. Tung and H. T. Choo, Macromol. Biosci., 2013, 13, 1092–1099 CrossRef CAS PubMed
- X. J. Loh, S. J. Ong, Y. T. Tung and H. T. Choo, Mater. Sci. Eng., C, 2013, 33, 4545–4550 CrossRef CAS PubMed
- X. J. Loh, J. Appl. Polym. Sci., 2013, 127, 992–1000 CrossRef CAS
- X. J. Loh, J. del Barrio, P. P. C. Toh, T. C. Lee, D. Z. Jiao, U. Rauwald, E. A. Appel and O. A. Scherman, Biomacromolecules, 2012, 13, 84–91 CrossRef CAS PubMed
- E. Y. Ye, P. L. Chee, A. Prasad, X. T. Fang, C. Owh, V. J. J. Yeo and X. J. Loh, Mater. Today, 2014, 17, 194–202 CrossRef CAS
- X. J. Loh, Mater. Horiz., 2014, 1, 185–195 RSC
- X. J. Loh, Abstracts of Papers of the American Chemical Society, 2014, p. 248 Search PubMed
- Q. Q. Dou, S. S. Liow, E. Y. Ye, R. Lakshminarayanan and X. J. Loh, Adv. Healthcare Mater., 2014, 3, 977–988 CrossRef CAS PubMed
- X. J. Loh, B. J. H. Yee and F. S. Chia, J. Biomed. Mater. Res., Part A, 2012, 100, 2686–2694 CrossRef PubMed
- X. J. Loh, W. Guerin and S. M. Guillaume, J. Mater. Chem., 2012, 22, 21249–21256 RSC
- V. P. N. Nguyen, N. Y. Kuo and X. J. Loh, Soft Matter, 2011, 7, 2150–2159 RSC
- X. J. Loh, P. N. N. Vu, N. Y. Kuo and J. Li, J. Mater. Chem., 2011, 21, 2246–2254 RSC
- M. Lotem, A. Hubert, O. Lyass, M. A. Goldenhersh, A. Ingber, T. Peretz and A. Gabizon, Arch. Dermatol., 2000, 136, 1475–1480 CAS
- M. Slingerland, H.-J. Guchelaar and H. Gelderblom, Drug Discovery Today, 2012, 17, 160–166 CrossRef CAS PubMed
- A. Chan, V. Shih and T. C. Kian, J. Oncol. Pharm. Pract., 2007, 13, 105–107 CrossRef CAS PubMed
- G. Stathopoulos, D. Antoniou, J. Dimitroulis, P. Michalopoulou, A. Bastas, K. Marosis, J. Stathopoulos, A. Provata, P. Yiamboudakis and D. Veldekis, Ann. Oncol., 2010, 21, 2227–2232 CrossRef CAS PubMed
- R. Suzuki, D. Omata, Y. Oda, J. Unga, Y. Negishi and K. Maruyama, Nanomaterials in Pharmacology, 2016, 457–482 Search PubMed
| This journal is © The Royal Society of Chemistry 2016 |