
The technology of straw sugar conversion to diol chemicals
Abstract
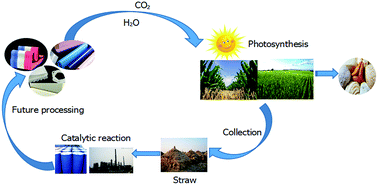
Straw sugar conversion to diols is one of the most favored routes for biomass utilization. In this paper, straw sugar was obtained by pretreating straw with ion exchange and decolorizing. The Sn–Co promoted RANEY® Ni catalyst for straw sugar hydrocracking to diols was prepared and characterized using BET, SEM, XRD, XPS and ICP. The yield of conversion of straw sugar to diols reached 50% under the optimum reaction conditions of 503 K at 11 MPa for 3 hours, with 15% catalyst content (solid–liquid mass ratio), 4 wt% alkali amount, pH 11.2–11.7, and 5 Nm3 h−1 hydrogen flow. The reaction mechanism was also hypothesized. This work may provide further guidance for the industrial production of diols from biomass.
 Please wait while we load your content...
Please wait while we load your content...

