
Metal-free C-3 alkylation of imidazopyridines with xanthates and convenient access to alpidem and zolpidem†
Abstract
A metal-free process for the C-3 alkylation of imidazopyridines has been developed. Various alkylation products including alkyl ester-, cyano-, ketone- and amide-substituted imidazopyridines were prepared in good yields. With this method, a highly efficient and concise formal synthesis of alpidem (A) and zolpidem (B) has been completed.
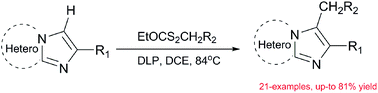
 Please wait while we load your content...
Please wait while we load your content...

