
Catalytic halodefluorination of aliphatic carbon–fluorine bonds†
Abstract
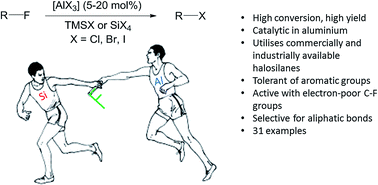
A variety of halosilanes, in conjunction with aluminum catalysts, convert fluorocarbons into higher halocarbons. Bromination and iodination of fluorocarbons are more effective than chlorination in terms of yield and activity. The mechanism for the reaction is investigated utilizing experimental and computational evidence and preliminary results suggest an alternate mechanism to that reported for the related hydrodefluorination reaction.
 Please wait while we load your content...
Please wait while we load your content...

