
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-stage synthesis of FcP(O)(OC2H5)2 from ferrocene and α-hydroxyethylphosphonate
Mikhail
Khrizanforov
,
Sofia
Strekalova
,
Kirill
Kholin
,
Vera
Khrizanforova
,
Valeriya
Grinenko
,
Tatyana
Gryaznova
and
Yulia
Budnikova
*
A.E. Arbuzov Institute of Organic and Physical Chemistry, Kazan Scientific Center of Russian Academy of Sciences, 8, Arbuzov str., 420088 Kazan, Russian Federation. E-mail: khrizanforov@gmail.com; Fax: +7 843 2732253; Tel: +7 843 2795335
First published on 25th April 2016
Abstract
A new approach is proposed for ferrocene phosphorylation using α-hydroxylalkylphosphonate as a “masked” phosphorylating agent, by electrochemical reduction of a ferrocene and (Me)2C(OH)P(O)(OC2H5)2 mixture at −50 °C. The method makes it possible to obtain the product of diethyl ferrocenyl phosphonate with a high yield (87–89%) and 100% conversion of the initial phosphonate in one stage. It is evidenced with experiments that ferrocene reduction is carried out with preservation of the iron charge in the ferrocene fragment and with the formation of a cyclopentadienyl ligand radical anion at −3.3 V ref. Ag/AgCl (at −50 °C).
Introduction
Ferrocene derivatives with phosphorus-containing substituents (phosphines, phosphonic acid moieties, etc.) gave rise to a new class of organophosphorus compounds, which are very powerful ligands that have unique features in metal-catalyzed organic reactions.1–15Ferrocene-bearing ligands proved to be prospective precursors in the design of various complexes and coordination polymers, which possess interesting and practically applicable physical features. The electrochemical, magnetic, luminescent and nonlinear optical features of coordination polymers with ferrocene fragments are of particular significance.3,7,16–20 This is due to the specific geometries that the ferrocenyl moiety can determine as well as to its electronic (redox) properties. Ferrocene can be regarded as a universal modifier for organic compounds and biomolecules. Due to stable sandwich structure and reversibility of oxidation under physiological pH, ferrocene can be used as a redox-active but nonradioactive label.21–24 Moreover, with its high lipophilicity, ferrocene easily penetrates cell and nuclear membranes and overcomes the blood brain barrier. Ferrocene as a carrier (or, in modern terms, a vector) can be used for the delivery of other molecules through membranes. It is important that the toxicity of organic compounds decreases upon Fc modification.24,25 Ferrocenyl organophosphorus ligands are usually stable, easy to handle and often isolable as optical stereoisomers. Ferrocenyl phosphine ligands are able to form complexes with transition metals in a variety of coordinative geometries and oxidation states, thus producing efficient catalyst precursors for many chemical transformations.1,16
The first attempts to introduce a phosphorous substituent to a ferrocene by means of (EtO)2P(O)H free-radical phosphorylation occurred as early as 1962;26 however, they were unsuccessful. It was shown that mixing a ferrocene suspension in phosphorous trichloride PCl3 with aluminum chloride AlCl3 and a subsequent hydrolysis of the reaction mixture would lead to the formation of ferrocenyl(H-phosphinic)acid and 1,1′-ferrocenyl-bis(H-phosphinic)acid with a low yield of 3–5%.27 The use of (dimethylamine)dichlorophosphine Me2NPCl2 as the phosphorylating reagent somehow made it possible to increase the yield of the acid to 9%.28 Ferrocenyl(phenyl)phosphonic acid was obtained with a high yield of 92% in the course of the reaction of ethyl ether of ferrocenyl(phenyl)phosphonic acid Fc(Ph)P(O)OEt with Me3SiBr and subsequent hydrolysis.29 However, Fc(Ph)P(O)OEt obtaining is based on the use of a hazardous gas Cl2, which makes this method much less attractive.
The most frequent method of synthesis of ferrocene derivatives with phosphorus-containing substituents is based on the metallation of ferrocene (for instance, with butyllithium) or its derivatives with a subsequent addition of chlorophosphine electrophile.16,18,29–33 Diethyl ferrocenyl phosphonate FcP(O)(OEt)2 was obtained with the yield of 33% using a reaction of ferrocene monolithium with diethylchlorophosphate (EtO)2P(O)Cl at 0 °C.27 The addition of a base tBuOK facilitated an increase in yield to 79% (at −78 °C in THF, Scheme 1).32
 | ||
| Scheme 1 Multi-stage approach to the synthesis of ferrocene derivatives. | ||
This approach is multi-stage, as it includes the use of t-BuLi (after adding tert-butoxide to ferrocene) at a very low temperature −78 °C at the first stage, and the use of a chlorinated derivative, i.e., diethyl chlorophosphate, which is toxic, unstable and results in chlorine-containing byproducts, at the second stage.32 It is also disadvantageous to use the explosive solvent THF. For that reason, such a method is expensive and environmentally unacceptable.
The latest achievements in the field of direct phosphorylation of the C(sp2)–H bons in a variety of aromatic substrates, including electrochemical functionalization, do not concern the phosphorylation of ferrocenes.34–37
Taking into account the importance of ferrocene derivatives with phosphor-containing substituents, search of new, more convenient, one-stage and selective approaches to their synthesis remains a relevant objective. The purpose of this work is to develop a method for the direct phosphorylation of ferrocene with α-hydroxylethylphosphonate under electrochemical reduction conditions and to identify the intermediate of this reaction.
Results and discussion
Electrochemical synthesis
A joint electrolysis of ferrocene and the phosphorylating agent α-hydroxylethylphosphonate (Me)2C(OH)P(O)(OC2H5)2 at −50 °C on a platinum electrode led to the formation of monosubstituted 1 (as the basic product) and disubstituted 2 diethyl ferrocenyl phosphonates FcP(O)(OEt)2 with good yields (Table 1). The electrolysis potential was −3.3 V (ref. to Ag/AgCl), and alkali were used as basic additives to activate the “masked” phosphorylating reagent.| No | Reagents and ratios | Base additive | Electrode/reaction potential | Producta yield |
|---|---|---|---|---|
a
 – 1-diethyl ferrocenyl phosphonate; 2-1,1′-ferrocenylbis-(diethylphosphonate); 3-ferrocenylphosphonic acid. – 1-diethyl ferrocenyl phosphonate; 2-1,1′-ferrocenylbis-(diethylphosphonate); 3-ferrocenylphosphonic acid.
|
||||
| 1 | Fc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :(CH3)2C(OH)P(O)(OEt)2 [1:1] :(CH3)2C(OH)P(O)(OEt)2 [1:1] |
Et4NOH | Pt/−3.3 V | 1, 88% |
| 2, 2% | ||||
| 2 | Fc:(CH3)2C(OH)P(O)(OEt)2 [1:2] |
Et4NOH | Pt/−3.3 V | 1, 60% |
| 2, 12% | ||||
| 3 | Fc:(CH3)2C(OH)P(O)(OEt)2 [1:1] |
NaOH + Et4NBF4 | Pt/−2.9 V | 1, 61% |
| 2, 6% | ||||
| 4 | Fc:HP(O)(OEt)2 [1:1] |
Et4NBF4 | Pb/−3.2 V | 1, 35% |
| 5 | Fc:H3PO3 [1:1] |
Et4NBF4 | Pb/−3.3 V | 3, 36% |
The use of Et4NOH provides higher yield compared to NaOH, probably because, in the latter case, due to side reactions of sodium reduction and the formation of sparingly soluble sodium salts of phosphorous acids. Alkaline conditions are necessary for a slow transformation of the “masked” phosphorylating agent into H-phosphonate by a known reaction,38–41 which we assumed would be quickly captured by the reduced form of ferrocene. In the absence of Et4NOH (or NaOH), phosphorylation with (CH3)2C(OH)P(O)(OEt2) does not occur.
Interestingly, in the proposed low-temperature conditions, phosphorous acid H3PO3 can also serve as a phosphorylating reagent using a lead cathode because it is not reduced in the available range of potentials on Pb at −50 °C (Table 1, line 5, product 3). An alkali activator is not required in this case. It should be noted that the lead cathode at low temperatures and high cathode potentials is gradually degraded, hence its use is limited to (by) only several cycles of synthesis. In this regard, preference was given to a platinum electrode and correspondingly suitable for it phosphorylated substrate, (Me)2C(OH)P(O)(OC2H5)2. After passing of 1F of electricity (not 2F, as shown in Table 1), the yield reduced by ca. 10–12%. Further increase (>2F) of the passed current led to a decrease in yield and an unidentified byproducts formation. Ferrocene phosphorylation reaction with the use of dialkyl-H-phosphonate at the lead cathode occurs non-selectively, the yield of target product, determined by 31P NMR spectra, was 35% (Table 1, line 4). Unidentified phosphorus-containing products were formed and the electrode was partially destroyed.
Voltammetry
Ferrocene and ferrocenyl derivatives are well known due to their ability to undergo a reversible one-electron oxidation,3,42 and the potential of Fc+/Fc is the internal standard for electrochemical measurement,43 the values of which have being measured in various aprotic and protonic solvents as well as in ionic liquids. Curiously, the reduction of ferrocene was studied in less detail, in single works that refer only to the voltammetry method.44–46A high-purity solvent and electrochemical measurements at temperatures ranging from −45 °C to −30 °C have made it possible to observe a reversible one-electron reduction of ferrocene on mercury, platinum and glassy-carbon electrodes in DMF and dimethoxyethane.45–47 However, the voltammograms are poorly reproducible, the approximate half-wave potential is −2.9 V (ref. electrode was not specified) at −37 °C (ref. 43) or −3.0 V at −90 °C in THF (ref. SCE).45 It was assumed that, as a result of the reduction, a relatively unstable Cp2Fe− anion would be formed.48 An increase in temperature leads to a two-electron reduction of the ferrocene and its decomposition, thereby forming metal iron on the mercury cathode.46 Attempts were made to reduce the ferrocene in the CO atmosphere, and the formation of [CpFe(CO)2]2 was observed.49
We have assumed the reversibility of the ferrocene reduction at low temperatures and, accordingly, the relative stability of the reduced form (at least, in the time scale of the voltammetry) could be used to perform the functionalization-phosphorylation with its participation. The choice of partner for the coupling the ferrocene is limited by its reduction potential; that is, its Ep should be more negative than that of the ferrocene, or it should be electrochemically inactive in the accessible cathode range of the potentials. The voltammograms of investigated phosphorylating agents reduction on electrodes with low and high hydrogen overvoltage (Pt and Pd, correspondingly) at 25 °C and −50 °C, respectively, are shown in Fig. 1. Earlier it was known that dialkyl phosphites were reduced on electrodes with low hydrogen overvoltage at room temperature on platinum or glassy-carbon type electrodes in aprotic solvent in the presence of ammonium salts as supporting electrolyte,50,51 although in the range of high potentials, but still comparable with that for ferrocene. We can clearly see that voltammograms of phosphorus compounds with P–H bonds ((EtO)2PHO, H3PO3) have no pronounced reduction peaks on platinum, as noted previously,51 but their electrochemical activity is reflected in the sharp decrease of the limiting potential at which current increases, apparently due to the discharge of phosphoric acids and background electrolyte. The evolution of hydrogen, which can be observed visually, leads to oscillations in the voltammograms. The use of a lead electrode with high hydrogen overvoltage allows to extend the available range of potentials, because no electrode reaction up to −3.4 V is observed (Fig. 1).
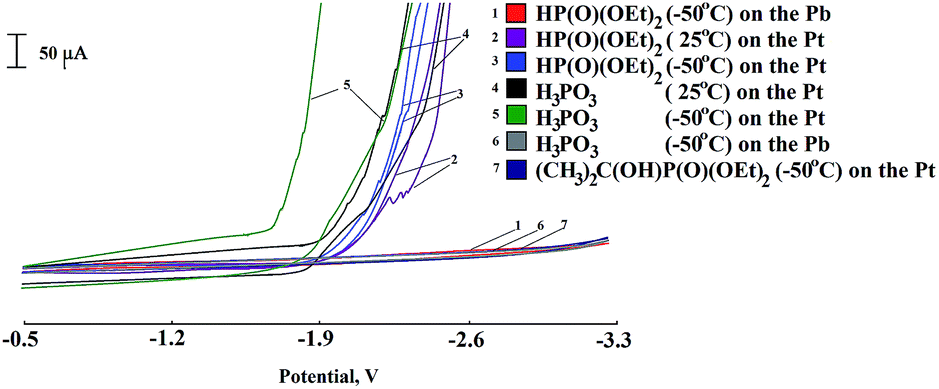 | ||
| Fig. 1 Cyclic voltammograms of phosphorylating agents. Conditions: T = 25 and −50 °C, working electrodes Pt (2 mm2) or Pb (16 mm2), Ag/AgCl ref. electrode, 50 mM concentration, Bu4NBF4, DMF, 100 mV s−1. | ||
Consequently, α-hydroxyethylphosphonate, so-called “masked” phosphorylating agent,38–41 is a more appropriate phosphorylating agent in our case (Table 1, lines 1–3). It is not reduced in the investigated accessible potential window in DMF on platinum cathode (Fig. 1) but is capable of slowly generating diethylphosphonate in the reaction mixture in the presence of a base.38–41
Cyclic voltammetry of ferrocene at the temperature of −50 °C is presented in Fig. 2 and Table 2. Accessible potential range increases at low temperatures. In the research conditions, two quasi-reversible peaks of oxidation and reduction of the ferrocene are observed, respectively. The quasi-reversible reduction at the potential of E1/2 = −3.10 V is well matched to the results of the ESR experiments, evidencing the formation of stable radical anions at this potential. The addition of a single electron to a neutral (uncharged) molecule generates chemical species called the radical anion (in our case, ferrocene radical-anion),52 which simultaneously has a unit of negative charge and an unpaired electron, as is known.
 | ||
| Fig. 2 Cyclic voltammogram of ferrocene. Conditions: T = −50 °C, glassy carbon working electrode, Ag/AgCl ref. electrode, 0.5 mM concentration, Bu4NBF4, DMF, 100 mV s−1. | ||
| Oxidation, Fc0/Fc+ | Reduction, Fc0/Fc˙− | ||||
|---|---|---|---|---|---|
| E ap/Ecp, V | ΔEa–cp, V | E 1/2, V | E cp/Eap, V | ΔEc–ap, V | E 1/2, V |
| 0.62/0.53 | 0.09 | 0.57 | −3.19/−3.01 | 0.18 | −3.10 |
There is no distinct reduction peak on the CV of the obtained diethylphosphonate at room temperature and at −50 °C in the available range of potentials. Selectivity of ferrocene phosphorylation, high yield of monophosphorylation product and trace amounts of by-products in the optimal synthesis conditions indicate that the products are stable in electrolysis conditions and do not undergo further redox or chemical reactions (Table 1). We investigated the electrochemical behavior of diethyl ferrocenyl phosphonate in the electrolysis conditions (Fig. 3, Table 3). It has been found that the CV of this product differs strongly from CV of an unsubstituted ferrocene. So, diethyl ferrocenyl phosphonate is characterized by quasi-reversible oxidation peak at more positive potentials compared to that for ferrocene, ΔEa–cp = 0.44 V (Table 3). Importantly, the phosphorylated ferrocene cathodic peak is not observed in the available potential range (Fig. 3). Apparently, heterogeneous rate constants of oxidation and reduction of the phosphorylated ferrocene is much less than those for ferrocene under similar conditions. The observed redox properties explain the success of the ferrocene phosphorylation.
 | ||
| Fig. 3 Cyclic voltammetry of diethyl ferrocenyl phosphonate. Conditions: T = −50 °C, glassy carbon working electrode, Ag/AgCl ref. electrode, 0.5 mM [Fe], Bu4NBF4 background salt, DMF, 100 mV s−1. | ||
| Oxidation, R-Fc0/R-Fc+ | Reduction, R-Fc0/R-Fc˙− | ||||
|---|---|---|---|---|---|
| E ap/Ecp, V | ΔEa–cp, V | E 1/2, V | E cp/Eap, V | ΔEc–ap, V | E 1/2, V |
| 0.94/0.50 | 0.44 | 0.72 | —/−2.95 | — | — |
ESR experiments
The key stage in the reaction under investigation is ferrocene reduction. Because up to now the conclusions about this process have been drawn only on the basis of voltammetry, we used modern research methods, namely, combined ESR – electrochemistry in order to clarify the key reduction product (and reagent in phosphorylation process). In the course of the ferrocene reduction in DMF at the temperature of −50 °C and the potential of −3.3 V, a signal was observed, the line width being equal to ΔHpeak–peak = 11 Gs and g = 2.000 (Fig. 4). It is known that, in the ground state, the ferrocene Fc0 is low-spin compound (D5h symmetry, 3d6, S = 0).53 It can be expected that, upon its reduction, a paramagnetic form Fc˙− should be formed, which has not previously been registered. However, the ESR spectra of radical anions of substituted ferrocenes were identified.54,55 Their g-factor values range from 2.003 to 2.028; the overall spectrum width varies from some units to tens of Gs; and the intensity of such spectra, as a rule, is not high. The low intensity can be caused by an inner-sphere reorganization of the ferrocene molecule, as assumed in the cited ref. 45 In this process, as well as in case of a neutral ferrocene, the spin state of the system probably changes. In all the substituted ferrocenes, the electron transfer to the Cp ligands occurs, and in some cases even splittings on hydrogen nuclei of one of the cyclopentadiene rings are registered.56 Often, as in our case, only a relatively wide single line with no splitting is observed. The lifetime of the paramagnetic product amounts to 1 to 2 minutes in the DMF solution, according to time of the spectrum disappearance after the potential has been removed from the working electrode. This coincides with the data on the lifetime of the ferrocene anion obtained previously.45 The foregoing makes it possible to suppose that the registered spectrum is attributable to the ferrocene radical anion. The g-factor value gives the evidence that, as in the case of substituted ferrocenes, the electron transfer occurs with participation of the orbital of the ligand but not iron. | ||
| Fig. 4 ESR signal of reduced ferrocene (at −3.3 V) in a DMF solution, −50 °C. g = 2.000, ΔHpeak–peak = 11 Gs. | ||
At further increasing negative potential of the electrode, the ESR spectra become more complicated (Fig. 5). It is most likely that they correspond to paramagnetic products of ferrocene molecule decomposition. The simulated signal sim1 with a splitting from two protons refers to a derivative of the free ligand without the ferrocene structure. The simulated signal sim2 most likely refers to a fragment with Fe. ESR signals exist for a long time after the potential has been disconnected, but their intensity did not reduce for at least half an hour at the temperature of −50 °C.
 | ||
| Fig. 5 ESR signals of reduced ferrocene at −3.9 V in a DMF solution at the temperature of −50 °C. sim1: g = 2.0063, 2: aH = 7 Gs ΔH = 0.7 Gs; sim2: g = 2.002, ΔHpeak–peak = 11 Gs. | ||
Reaction scheme
The following Scheme 2 can be proposed for the electrochemical synthesis of phosphorylated ferrocene: | ||
| Scheme 2 Electrochemical synthesis of ferrocene derivative. | ||
In fact, in the reaction conditions we simultaneously generated dialkyl H-phosphonate from (Me)2C(OH)P(O)(OC2H5)2 by a known reaction, as well as ferrocene radical anions, which react with each other, ultimately producing a phosphorylated ferrocene with good yield in a single stage. The proposed scheme is the best possible, but it does not preclude any other separate stage that might lead to the same final product.
Conclusions
Thus, the ferrocene radical anion ESR spectrum was recorded during Fc electrochemical reduction at the temperature of −50 °C for the first time. We proposed new approach to the synthesis of phosphorylated ferrocene in one stage using α-hydroxylethylphosphonate and phosphorous acid as phosphorylating agents with the total yield up to 90%, and 36%, respectively.Experimental
Cyclic voltammetry
The BASi Epsilon E2P (USA) electrochemical workstation was used for voltammetric measurements. The device comprises a measuring unit, a Dell Optiplex 320 computer with Epsilon-EC-USB-V200 software. As background electrolytes, tetrafluoroborate of tetrabutylammonium (C4H8)4NBF4 was used. The working electrode was a stationary disc glassy-carbon electrode (the surface area of 6 mm2). Ag/AgCl (0.01 M) was used as a reference electrode, which was linked to the solution on the cell using a modified Luggin capillary filled with the background electrolyte (0.1 M Bu4NBF4 in DMF). Thus, the assembled electrode had two sections, each of which was completed with an ultra-thin frit glass (Vycor) to divide the AgCl from the analyzed compound.A platinum wire was used as an auxiliary electrode. The curve recording was performed at the potential linear sweep rate of 100 mV s−1. The measurements were performed in a temperature-controlled electrochemical cell (volume from 1 ml to 5 ml) in an inert gas atmosphere (N2). The cooling of the researched solutions was performed with frozen carbon dioxide.
Between measurements or prior to the registration of a voltammetry wave, the solution was actively stirred with a magnetic stirrer in the atmosphere of constant inflow of an inert gas that was run through a dehydrating system, and then through a BI-GAScleaner (manufactured by OOO Modern Laboratory Equipment, Novosibirsk) nickel-based purification system to remove any trace quantity of oxygen.
ESR research
Liquid samples for the ESR spectra registration were freed from oxygen by applying a three-time repeat of the following cycle: freezing in liquid nitrogen, pumping and defrosting. After the last cycle, the electrolysis cell was filled with gaseous helium. The operating electrode was a gold spiral. The experiments were performed in DMF at −50 °C, the background electrolyte was 0.1 M Bu4NBF4, and the sweep rate E(t) amounted to 0.1 V s−1. The measurements were performed with a suite of hardware and software assembled on the basis of the electrochemical analogue with a potentiostat and a PWR-3 programmer, an ELEXSYS E500 X-range ESR spectrometer, an E14-440 ADC and DAC module (L-Card Company), a computer and an original three-electrode spiral cell.57 The ESR spectra were simulated with the WinSim 0.96 (NIEHS) software.Preparative electrochemical syntheses
All reactions were obtained in a dry argon atmosphere. The preparative electrolysis was performed using a direct current source B5-49 in a three-electrode cell with 40 ml volume with a separation of the anode and cathode compartments. The potential value of the working electrode was recorded using a direct-current V7-27 voltmeter in relation to the Ag/AgCl (0.01 M, NaCl) reference electrode that had two sections separated with Vycor, the second of which contained a saturated solution of the background salt in DMF. The surface area of the working platinum (Pt) U-shaped electrode amounted to 48.00 cm2. A ceramic plate with the pore rate of 900 nm was used as a membrane. During the preparative synthesis, the electrolyte was continuously stirred using a magnetic stirrer with a continuous inflow of inert gas that was run through a purification system in order to remove any traces of oxygen and other gaseous impurities.NMR spectra were recorded with a Bruker AVANCE-400 multi-nuclear spectrometer (400.1 MHz (1H), 100.6 MHz (13C) and 162.0 MHz (31P)). Chemical shifts are given in parts per million relative to SiMe4 (1H, internal solvent) and 85% H3PO4 (31P, external).
Reagents and research subjects
Dimethylformamide (“extra pure” by Acrosorganics), was purified by means of double fractionation distillation over melting potash. Diethyl(2-hydroxypropan-2-yl)phosphonate was obtained through the method described in the literature.58Et4NBF4 was obtained by mixing 30–35% water solution of tetraethylammonium hydroxide, Et4NOH and HBF4 acid to a neutral indicator reaction. In the course of the reaction, a white crystal precipitation is deposited, which is filtered and dehydrated. The obtained powder salts were recrystallized from ethyl ester and were dried for 2 to 3 days in a vacuum at 55 °C.
Ferrocene (98%) and H3PO3 (extra pure, 98%, Acrosorganics) were used without preliminary purification.
Et4NOH (20% aqueous solution, Acrosorganics) was subjected to a complete dehydration to a white solid on Schlenk's system prior to the beginning of the experiment.
General method of ferrocene phosphorylation
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1052 (P–O). EIMS, m/z: 322.15 [M]+; anal. calc.: C, 52.20; H, 5.95; Fe, 17.34; O, 14.90; P, 9.62; for C14H19FePO3; found: C, 52.14; H, 5.91; P, 9.58, Fe, 17.24. The spectroscopic data for diethyl ferrocenyl phosphonate matched that reported in the literature.28
O), 1055 (P–O). EIMS, m/z: 458.57 [M]+; anal. calc.: C, 47.18; H, 6.16; Fe, 12.19; O, 20.95; P, 13.52 for C18H28FeO6P2; found: C, 47.16; H, 6.09, P, 13.45, Fe, 12.13. The spectroscopic data for 1,1′-ferrocenylbis(diethylphosphonate) matched that reported in the literature.28
O), 1030 (P–O). EIMS, m/z: 266.70 [M]+; anal. calc.: C, 45.11; H, 4.13; P, 11.65, Fe, 20.97. for C10H11FePO3 found: C, 45.16; H, 4.10; P, 11.69, Fe, 20.60. The spectroscopic data for ferrocenylphosphonic acid matched that reported in the literature.28
O), 1052 (P–O). EIMS, m/z: 322.15 [M]+; anal. calc.: C, 52.20; H, 5.95; Fe, 17.34; O, 14.90; P, 9.62; for C14H19FePO3; found: C, 52.14; H, 5.91; P, 9.58, Fe, 17.24. The spectroscopic data for diethyl ferrocenyl phosphonate matched that reported in the literature.28
O), 1055 (P–O). EIMS, m/z: 458.57 [M]+; anal. calc.: C, 47.18; H, 6.16; Fe, 12.19; O, 20.95; P, 13.52 for C18H28FeO6P2; found: C, 47.16; H, 6.09, P, 13.45, Fe, 12.13. The spectroscopic data for 1,1′-ferrocenylbis(diethylphosphonate) matched that reported in the literature.28
O), 1030 (P–O). EIMS, m/z: 266.70 [M]+; anal. calc.: C, 45.11; H, 4.13; P, 11.65, Fe, 20.97. for C10H11FePO3 found: C, 45.16; H, 4.10; P, 11.69, Fe, 20.60. The spectroscopic data for ferrocenylphosphonic acid matched that reported in the literature.28
Acknowledgements
The work was supported with a grant from the Russian Science Foundation No. 14-23-00016.Notes and references
- P. Barbaro, C. Bianchini, G. Giambastiani and S. L. Parisel, Coord. Chem. Rev., 2004, 248, 2131 CrossRef CAS.
- T. J. Colacot, Chem. Rev., 2003, 103, 3101 CrossRef CAS PubMed.
- A. Togni and T. Hayashi, Ferrocenes: homogeneous catalysis, organic synthesis, material science, Wiley-VCH, Weinheim, 1995 Search PubMed.
- H. B. Kagan, P. Diter, A. Gref, D. Guillaneux, A. Masson-Szymczak, F. Rebière, O. Riant, O. Samuel and S. Taudien, Pure Appl. Chem., 1996, 68, 29 CrossRef CAS.
- C. J. Richards and A. J. Locke, Tetrahedron: Asymmetry, 1998, 9, 2377 CrossRef CAS.
- R. C. J. Atkinson, V. C. Gibson and N. J. Long, Chem. Soc. Rev., 2004, 33, 313 RSC.
- P. Stepnicka, Ferrocenes: ligands, materials and biomolecules, Chichester, 2008 Search PubMed.
- A. Togni, N. Bieler, U. Burckhardt, C. Köllner, G. Pioda, R. Schneider and A. Schnyder, Pure Appl. Chem., 1999, 71, 1531 CrossRef CAS.
- L.-X. Dai, X.-L. Hou, W.-P. Deng, S.-L. You and Y.-G. Zhou, Pure Appl. Chem., 1999, 71, 1401 CAS.
- L.-X. Dai, T. Tu, S.-L. You, W.-P. Deng and X.-L. Hou, Acc. Chem. Res., 2003, 36, 659 CrossRef CAS PubMed.
- T. J. Colacot, Chem. Rev., 2003, 103, 3101 CrossRef CAS PubMed.
- X. L. Hou, S. L. You, T. Tu, W. P. Deng, X. W. Wu, M. Li, K. Yuan, T. Z. Zhang and L. X. Dai, Top. Catal., 2005, 35, 87 CrossRef CAS.
- R. Gómez Arrayás, J. Adrio and J. C. Carretero, Angew. Chem., Int. Ed., 2006, 45, 7674 CrossRef PubMed.
- M. Ritte, C. Bruhn and U. Siemeling, Z. Naturforsch., B: J. Chem. Sci., 2014, 69, 906 CAS.
- M. Neel, A. Panossian, A. Voituriez and A. Marinetti, J. Organomet. Chem., 2012, 716, 187 CrossRef CAS.
- D. Astruc, in Organometallic Chemistry and Catalysis, Springer-Verlag, Berlin, 2007 Search PubMed.
- M. N. Khrizanforov, D. M. Arkhipova, R. P. Shekurov, T. P. Gerasimova, V. V. Ermolaev, D. R. Islamov, V. A. Miluykov, O. N. Kataeva, V. V. Khrizanforova, O. G. Sinyashin and Y. H. Budnikova, J. Solid State Electrochem., 2015, 19, 2883 CrossRef CAS.
- D. Schaarschmidt and H. Lang, Organometallics, 2013, 32, 5668 CrossRef CAS.
- W.-Y. Wang, N.-N. Ma, L. Wang, C.-L. Zhu, X.-Y. Fang and Y.-Q. Qiu, Dyes Pigm., 2016, 126, 29 CrossRef CAS.
- V. Mereacre, M. Schlageter, A. Eichhöfer, T. Bauer, J. A. Wolny, V. Schünemann and A. K. Powell, J. Magn. Magn. Mater., 2016, 407, 87 CrossRef CAS.
- S. Takenaka, Y. Uto, H. Saita, M. Yokoyama, H. Kondo and W. D. Wilson, Chem. Commun., 1998, 1111 RSC.
- S. Sato and S. Takenaka, J. Organomet. Chem., 2008, 693, 1177 CrossRef CAS.
- S. Watanabe, S. Sato, K. Ohtsuka and S. Takenaka, Anal. Chem., 2011, 83, 7290 CrossRef CAS PubMed.
- L. V. Snegur, A. A. Simenel, A. N. Rodionov and V. I. Boev, Russ. Chem. Bull., 2014, 63, 26 CrossRef CAS.
- L. V. Snegur, Y. S. Nekrasov, N. S. Sergeeva, Z. V. Zhilina, V. V. Gumenyuk, Z. A. Starikova, A. A. Simenel, N. B. Morozova, I. K. Sviridova and V. N. Babin, Appl. Organomet. Chem., 2008, 22, 139 CrossRef CAS.
- E. F. Jason and E. K. Fields, J. Org. Chem., 1962, 27, 1402 CrossRef CAS.
- G. P. Sollott and E. Howard, J. Org. Chem., 1962, 27, 4034 CrossRef CAS.
- G. R. Knox, P. L. Pauson and D. Willison, Organometallics, 1992, 11, 2930 CrossRef CAS.
- O. Oms, J. Le Bideau, A. Vioux and D. Leclercq, J. Organomet. Chem., 2005, 690, 363 CrossRef CAS.
- M. Korb, D. Schaarschmidt and H. Lang, Organometallics, 2014, 33, 2099 CrossRef CAS.
- S. R. Alley and W. Henderson, J. Organomet. Chem., 2001, 216, 637 Search PubMed.
- O. Oms, F. Maurel, F. Carré, J. Le Bideau, A. Vioux and D. Leclercq, J. Organomet. Chem., 2004, 689, 2654 CrossRef CAS.
- R. P. Shekurov, V. A. Miluykov, D. R. Islamov, D. B. Krivolapov, O. N. Kataeva, T. P. Gerasimova, S. A. Katsyuba, G. R. Nasybullina, V. V. Yanilkin and O. G. Sinyashin, J. Organomet. Chem., 2014, 766, 40 CrossRef CAS.
- Y. H. Budnikova and O. G. Sinyashin, Russ. Chem. Rev., 2015, 84, 917 CrossRef CAS.
- T. Gryaznova, Y. Dudkina, M. Khrizanforov, O. Sinyashin, O. Kataeva and Y. Budnikova, J. Solid State Electrochem., 2015, 19, 2665 CrossRef CAS.
- T. V. Gryaznova, Y. B. Dudkina, D. R. Islamov, O. N. Kataeva, O. G. Sinyashin, D. A. Vicic and Y. H. Budnikova, J. Organomet. Chem., 2015, 785, 68 CrossRef.
- M. Khrizanforov, S. Strekalova, T. Gryaznova, V. Khrizanforova and Y. Budnikova, Russ. Chem. Bull., 2015, 8, 1926 CrossRef.
- C. Li, T. Yano, N. Ishida and M. Murakami, Angew. Chem., 2013, 125, 9983 CrossRef.
- D. W. Allen, D. E. Hibbs, M. B. Hursthouse and K. M. A. Malik, J. Organomet. Chem., 1999, 572, 259 CrossRef CAS.
- G. A. Stark, T. H. Riermeier and M. Beller, Synth. Commun., 2000, 30, 1703 CrossRef CAS.
- M. Hayashi, T. Matsuura, I. Tanaka, H. Ohta and Y. Watanabe, Org. Lett., 2013, 15, 628 CrossRef CAS PubMed.
- N. G. Tsierkezos, J. Solution Chem., 2007, 36, 289 CrossRef CAS.
- R. R. Gagné, C. A. Koval and G. C. Lisensky, Inorg. Chem., 1980, 19, 2854 CrossRef.
- N. Ricke, S. N. Eustis and K. H. Bowen, J. Mass Spectrom., 2014, 357, 63 CAS.
- N. Ito, T. Saji and S. Aoyagui, J. Organomet. Chem., 1983, 247, 301 CrossRef CAS.
- V. V. Strelets, Coord. Chem. Rev., 1992, 114, 1 CrossRef CAS.
- Y. Mugnier, C. Moise, J. Tirouflet and E. Laviron, J. Organomet. Chem., 1980, 186, 49 CrossRef.
- A. J. Bard, E. Garcia, S. Kukbarenko and V. V. Strelets, Inorg. Chem., 1993, 32, 3528 CrossRef CAS.
- N. E. Murr and A. Chaloyard, J. Organomet. Chem., 1980, 193, 60 CrossRef.
- Y. H. Budnikova, T. R. Novoselova and Y. M. Kargin, Russ. J. Gen. Chem., 1992, 62, 19621 Search PubMed.
- A. P. Tomilov, B. I. Martynov and N. A. Pavlova, J. Electroanal. Chem., 2001, 507, 46 CrossRef CAS.
- A. Alberti, M. Benaglia, B. F. Bonini, M. Fochi, D. Macciantelli, M. Marcaccio, F. Paolucci and S. Roffia, J. Phys. Org. Chem., 2004, 17, 1084 CrossRef CAS.
- T. P. Gryaznova, S. A. Katsyuba, V. A. Milyukov and O. G. Sinyashin, J. Organomet. Chem., 2010, 695, 2586 CrossRef CAS.
- C. Elschenbroich and M. Cais, J. Organomet. Chem., 1969, 18, 135 CrossRef CAS.
- G. Bigam, J. Hooz, S. Linke, R. E. D. McClung, M. W. Mosher and D. D. Tanner, Can. J. Chem., 1972, 50, 1825 CrossRef CAS.
- E. V. Tretyakov, S. E. Tolstikov, G. V. Romanenko, A. S. Bogomyakov, D. V. Stass, M. K. Kadirov, K. V. Holin, O. G. Sinyashin and V. I. Ovcharenko, Polyhedron, 2011, 30, 3232 CrossRef CAS.
- M. K. Kadirov, E. V. Tret'yakov, Y. G. Budnikova, K. V. Kholin, M. I. Valitov, V. N. Vavilova, V. I. Ovcharenko, R. Z. Sagdeev and O. G. Sinyashin, Russ. J. Phys. Chem. A, 2009, 83, 2163 CrossRef CAS.
- F. Texier-Boullet and M. Lequtte, Tetrahedron Lett., 1986, 27, 3515 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |