
Sorption discrimination between secondary alcohol enantiomers by chiral alkyl-dicarboxylate MOFs†
Abstract
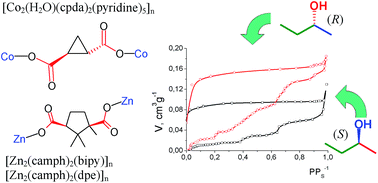
The 3D coordination polymer [Co2(H2O)(cpda)2(py)4·py]n (cpdaH2 is trans-(S,S)-1,2-cyclopropane dicarboxylic acid, py = pyridine) crystallizes from pyridine as 1·5py (one py is not coordinated) and was characterized by X-ray single crystal diffraction. Desolvation of 1·5py was accomplished with decoordination of pyridine and transformation of the CoII octahedral coordination into tetrahedral, as confirmed by electronic spectroscopy. Sorption of individual optical isomers – (S)-2-butanol and (R)-2-butanol – from the gas phase at 303 K by desolvated 1 was studied, and for comparison sorption of these substrates by the chiral MOFs [Zn2(camph)2(bipy)]n (2) and [Zn2(camph)2(dpe)]n (3) was examined (camphH2 is (1R,3S)-camphoric acid, bipy is 4,4′-bipyridine, dpe is trans-1,2-di(4-pyridyl)ethylene). Chiral sites in 1–3 contain only one polar group (carboxylate) in close proximity to the asymmetric C atom, while the other groups contain only C–H or C–C bonds. In the cases of 1 or 2 the absorption isotherms grew abruptly at certain pressure values P, and these values were different for the (R) or (S) isomers' sorptions. Such differential growth can be accounted for through the polymeric framework's rearrangement induced by interaction with 2-butanol, the difference in P values for (R) and (S) isomers being an indication of different interaction energies for these isomers with the MOF. There was no significant difference between the values of total sorption capacity of 1 for the two enantiomers of 2-butanol at pressures close to the saturation vapor pressure. In contrast, the sorption capacity of 3 was higher for (R)-2-butanol than for (S)-2-butanol over the whole pressure range.
 Please wait while we load your content...
Please wait while we load your content...

