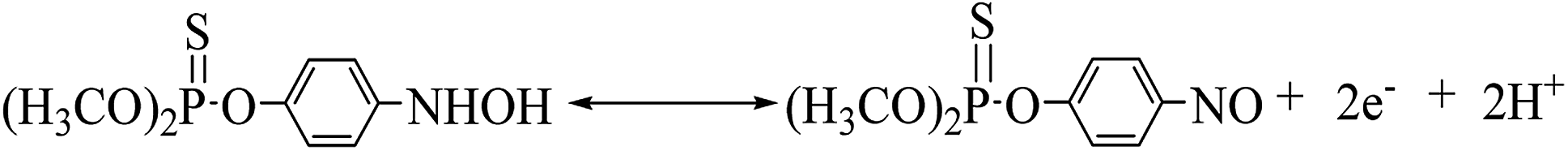DOI:
10.1039/C6RA09335C
(Paper)
RSC Adv., 2016,
6, 58771-58779
Facile and sensitive electrochemical detection of methyl parathion based on a sensing platform constructed by the direct growth of carbon nanotubes on carbon paper
Received
11th April 2016
, Accepted 12th June 2016
First published on 13th June 2016
Abstract
This work developed an economic, convenient, sensitive and practical methyl parathion sensor using an easy-to-fabricate, reusable, inexpensive carbon nanotube/carbon paper composite as the working electrode. Carbon nanotubes form a network structure and provide large surface area, which can enhance the electron transfer capability of the carbon paper electrode. As an electrochemical sensing platform, the constructed carbon nanotube/carbon paper sensor allows sensitive determination of methyl parathion in the range of 10 ng mL−1 to 1000 ng mL−1 with a low detection limit of 3.9 ng mL−1. Importantly, the present study demonstrated the high sensitivity, satisfactory stability and good anti-interference capabilities of this new electrode platform for methyl parathion detection. Furthermore, the present method was successfully applied to determine methyl parathion in kiwi samples with satisfactory precision (3.74–5.05%) of relative standard deviation (RSD) and acceptable recoveries (99.67–108%), which demonstrated the applicability of the carbon nanotube/carbon paper composite sensor. Such a facile approach for the detection of OPs may provide some guidance for designing nanomaterial-based sensors.
1. Introduction
Organophosphorus pesticides (OPs) are one of the most widely used pesticide types for pest control in agriculture.1,2 It is well-known that OPs can disrupt the cholinesterase enzyme that regulates acetylcholine, a neurotransmitter needed for proper nervous system function and lead to headache, dizziness, vomiting, loss of coordination and even death.3 Owing to their persistence, bioaccumulation and toxicity, OPs may cause long-term damage to the environment and living species even if their concentration is very low.4 Methyl parathion (MP) as an OP has been largely used in many countries to avoid agriculture losses caused by insect attacks. So far, numerous analytical methods such as gas chromatography,5–7 HPLC8 and capillary electrophoresis9,10 have been widely used for the determination of MP.11 Although accurate and sensitive, these techniques are usually expensive, laborious, time-consuming and not convenient for on-site or in-field detection.12 Hence, the establishment of a sensitive, rapid and convenient method to identify and quantify trace MP in various samples has gained significance in food safety, human health and environmental protection.
Electrochemical methods are simple in their setup as well as in the operation and data acquisition, so considerable efforts have been made to apply electrochemical analysis to achieve ultrasensitive and rapid detection of OPs.1,13–15 In general, electrochemical analysis methods used in the qualitative determination of OPs include enzymatic methods and enzyme-free methods.16 But, the denaturation of enzymes will result in the instability and short lifetime of use for the sensor.17 Moreover, enzymatic methods involved a multiple-step operation involving time-consuming incubation and reactivation/regeneration processes. The above factors limit wide applications of biosensors based the enzyme.2 Therefore, to eliminate the influence of enzyme on the analysis of OPs, some more effective methods were proposed to directly detect OPs without using enzymes in food and environmental samples.18–20
Remarkably, electrochemical voltammetric techniques attracted more and more attention for on-site monitoring of methyl parathion in the past decades. Su's group used the indium tin oxide electrodes modified with a highly ordered and aligned binary assembly of silica mesochannels and micelles to analyze of trace nitroaromatic explosives and OPs by differential pulse voltammetry (DPV), which showing a fast response, wide linear range, high sensitivity, and low detection limit at the ppb level.21 Tang and his partners constructed a methyl parathion sensor based on carbon nanotube and ionic liquid. The electrochemical detection of methyl parathion was achieved by linear sweep voltammetry (LSV) in the range from 0.1 to 2.5 μg mL−1 with a detection limit of 8 ng mL−1.22 MP can be determined in the range of 0.1 to 10 μg mL−1 with a detection limit of 0.01 μg mL−1 by cyclic voltammetry (CV) with a molecularly imprinted sensor based on nitrogen doped graphene sheets.23 In addition, a few of electrochemical sensors based on carbon nanomaterial and noble metal hybrids such as gold nanoparticles decorated carbon nanotubes nanocomposites,24–26 gold nanoparticles decorated graphene hybrids14 and other carbon nanomaterial based hybrids22,27–29 have also been widely used in the determination of methyl parathion by voltammetric techniques, which exhibits superior sensing properties of MP. Although these sensors exhibit excellent analytical performance, most of them are constructed on the basis of commercial electrodes, such as glassy carbon electrode and gold electrode, which always involve tedious pretreatment procedures of electrode cleaning and polishing, and need further multiple modifications process by nanomaterials. Therefore, it's necessary to exploit new, convenient and diversifying electrodes to develop easy-to-fabricate and inexpensive electrochemical sensors.
With the development of nanotechnology, nanomaterial has been applied in the construction of electrochemical sensors to improve their analytical performance. Among numerous nanomaterial, CNTs offer promising applications in fields ranging from electrochemistry to biotechnology because of their outstanding physical, electrical and thermal properties.16,30 Non-enzymatic electrochemical sensors based on CNTs have been used in the fabrication of OPs electrochemical sensors such as Pd/MWCNTs nanocomposite12 and Au/CNTs modified electrode.18,31 Although these sensors have good performance, they all involve dispersion of CNTs, complex modification process and noble metal of high-cost. Recently, CNTs directly grown onto electrically conductive substrates such as metal substrates and carbon cloth via chemical vapor deposition (CVD) method have been developed and applied immediately as electrodes for fuel cells, supercapacitors and electrocatalysis.32–36 However, CNTs grown on carbon paper (CP) directly used as the working electrode for electrocatalysis has been rarely reported.
In this work, using an easy-to-fabricate, reusable, inexpensive carbon nanotubes/carbon paper composite as the working electrode, we present a very simple electrochemical sensing platform that allows sensitive and selective determination of methyl parathion in real samples. This sensing platform shows a wide linear concentration ranges (10–1000 ng mL−1), a low detection limit (3.9 ng mL−1), and excellent reproducibility, stability and anti-interference. This simple sensor could potentially be produced in a large scale, providing a promising alternative for on-site detection of MP and other potential ingredients.
2. Experimental
2.1. Materials and reagents
MP was purchased from China National Institute of standardization methanol, ethanol, nickel acetate and other reagent were obtained from Sinopharm Chemical Reagent Co. Ltd. CP was obtained from Zhuzhou Chenxin Induction Equipment Co. Ltd. All the chemicals were of analytical reagent grade and used without further purification. Distilled water was used throughout the experiments. All experiments were carried out at ambient temperature.
2.2. Synthesis of CNTs/CP composite
CNTs/CP composite was prepared by CVD method. Detailed content is as follows: 10 mM nickel acetate ethanol solution as catalyst was spin-coated onto carbon paper substrate using a commercial spin-coater at a speed of 3000 rpm. Chemical vapor deposition was carried out using a commercially available thermal CVD system. The carbon paper substrate was placed in a tube furnace and annealed in an argon/hydrogen (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) environment at 900 °C to ensure that Ni was in their reduced state. Acetylene gas was introduced into the chamber at a rate of 200 mL min−1, 900 °C for 10 min during the growth step. Meanwhile, the furnace temperature was then kept at 900 °C for 30 min, which reduced the nickel(III) to nickel nanoparticles. After the growth, both hydrogen and acetylene (4:1) gases were turned off and substrates subsequently cooled to room temperature in argon/hydrogen environment. Afterwards, CNT/CP composites were obtained. Before electrochemical tests, CP electrode or CNTs/CP composite was tailored to proper size for use.
:5) environment at 900 °C to ensure that Ni was in their reduced state. Acetylene gas was introduced into the chamber at a rate of 200 mL min−1, 900 °C for 10 min during the growth step. Meanwhile, the furnace temperature was then kept at 900 °C for 30 min, which reduced the nickel(III) to nickel nanoparticles. After the growth, both hydrogen and acetylene (4:1) gases were turned off and substrates subsequently cooled to room temperature in argon/hydrogen environment. Afterwards, CNT/CP composites were obtained. Before electrochemical tests, CP electrode or CNTs/CP composite was tailored to proper size for use.
2.3. Electrochemical measurements
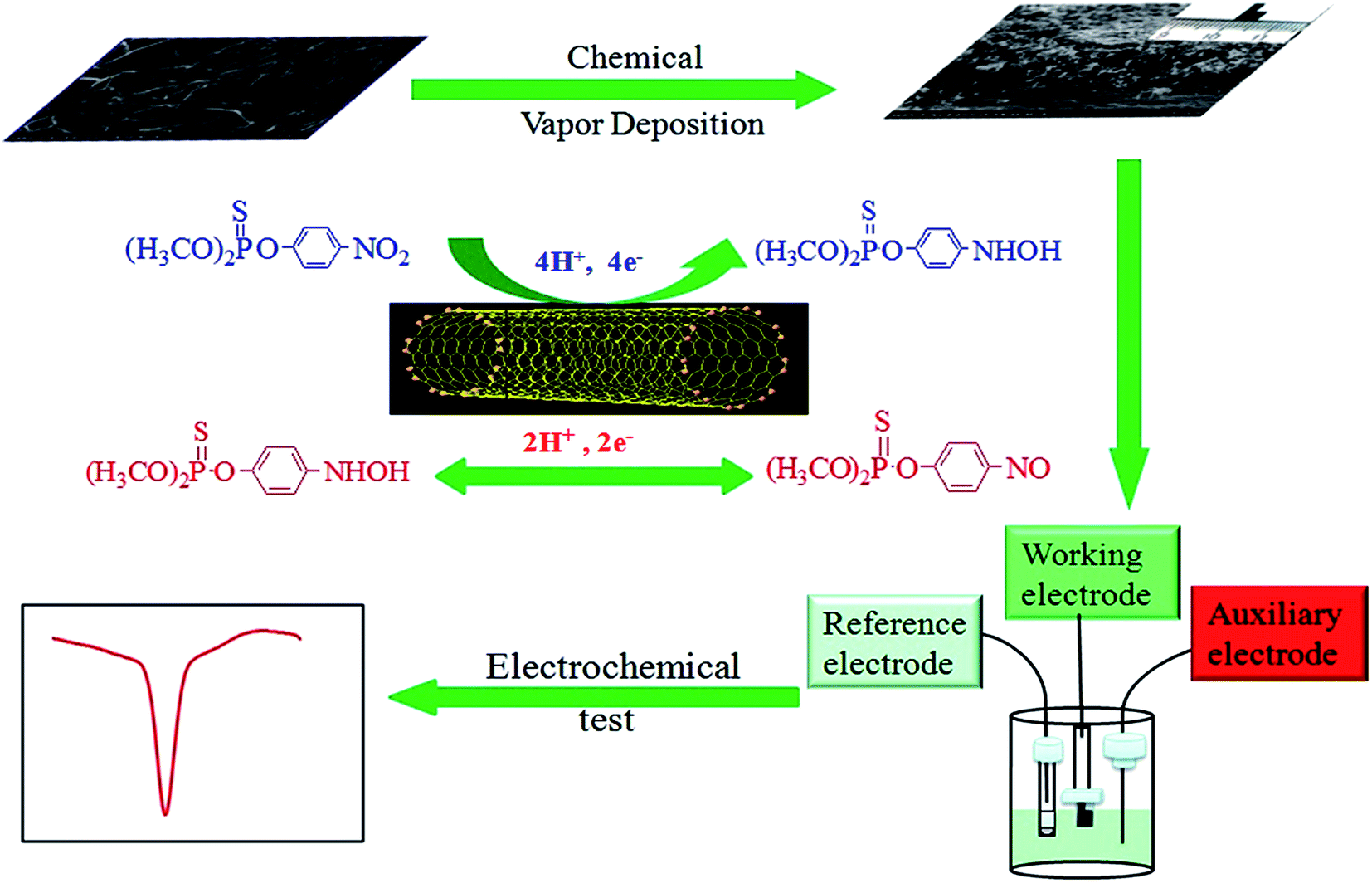
The procedure for the electrochemical sensor fabricated by CVD is illustrated in Scheme 1. For the adsorption procedure, the CNTs/CP composite was immersed into the acetate buffer solution containing the desired concentration of MP for a given time. Electrochemical tests were performed in 0.2 M acetate buffer solution (pH 5.0). Prior to each test, the solution were deoxygenated by purging with argon for 15 min. Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were performed on a CHI 630E electrochemical workstation (CHI Instruments) with a conventional three-electrode system comprised of a platinum wire as auxiliary electrode, a Ag/AgCl electrode (in 3 M KCl solution) as reference electrode, and the prepared CNTs/CP composite as working electrode, respectively.
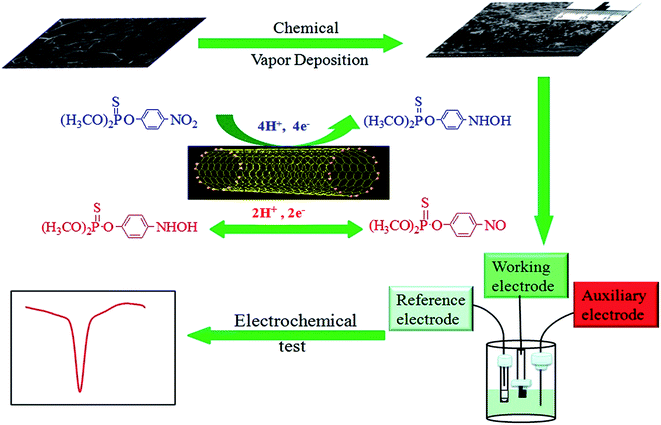 |
| | Scheme 1 Schematic diagram of the procedure for the electrochemical sensor fabricated by CVD. | |
2.4. Sample pretreatment
Kiwi samples were purchased from a local supermarket and prepared as follows: kiwi fruit samples were firstly peeled and a certain amount of peeled samples was weighed, then sprayed with a known amount of MP except the blank samples. 25 grams of peeled kiwi samples and 50.0 mL acetonitrile were placed in the refiner and homogenized for 2 min and filtered through filter paper. Filtrate was collected a scale test tube with 5 g NaCl, tucked the test tube with a plug and vigorously shocked for 1 min. After a few minutes' standing, the layering appeared between two-phase. Then 10 mL mixture was put in a beaker and evaporated to dryness. 1 mL acetone was used to wash the dry residue. The prepared solution for electrochemical determination was diluted with 0.2 M pH 5.0 acetate buffer solution, enriched for 300 s and tested by electrochemical method.
3. Results and discussion
3.1. Physical characterization
The SEM image shows that the carbon paper we used has a lamellar structure (Fig. 1A) and the thickness of which was about 0.11 mm measured by micrometer. Generally, SWCNTs have a small diameter (from 0.4 nm to 4 nm) and the diameter of a MWCNT can range from 1 nm to 100 nm. Thus, the prepared carbon nanotubes with the diameter of 50–100 nm should be multiwalled. In addition, carbon nanotubes that we prepared have a three-dimensional network structure (Fig. 1B) with the outer diameters of CNTs being 50–100 nm, the lengths of CNTs being larger than 1 μm and the thickness of CNTs layer being 10 μm. High specific surface area and well-conducting networks of CNTs can make CNTs/CP composite yield an excellent electron transfer pathway. According to the literatures by Wang Chao, it's predicted that the maximum interaction forces between the CNTs and CP may be decomposed into two components: the maximum pulling force and maximum lateral force, which are due to the van der Waals (vdWs) and chemical bonding interaction. Thus, the possible interaction between carbon paper and carbon nanotubes is attributed to the van der Waals (vdWs) and chemical bonding interaction.37 The TEM image exhibits typical structure of multiwalled carbon nanotubes (Fig. 1C), confirming the formation of carbon nanotubes but not carbon nanofibers in our experiment. Moreover, the composite can be cut into any shape according to desired requirement just as described in the inset of Fig. 1B.
 |
| | Fig. 1 (A) SEM of carbon paper; (B) SEM profile of CNTs/CP composite; inset: photograph of CNTs/CP electrode for electrochemical application; (C) TEM of CNTs grown on CP. | |
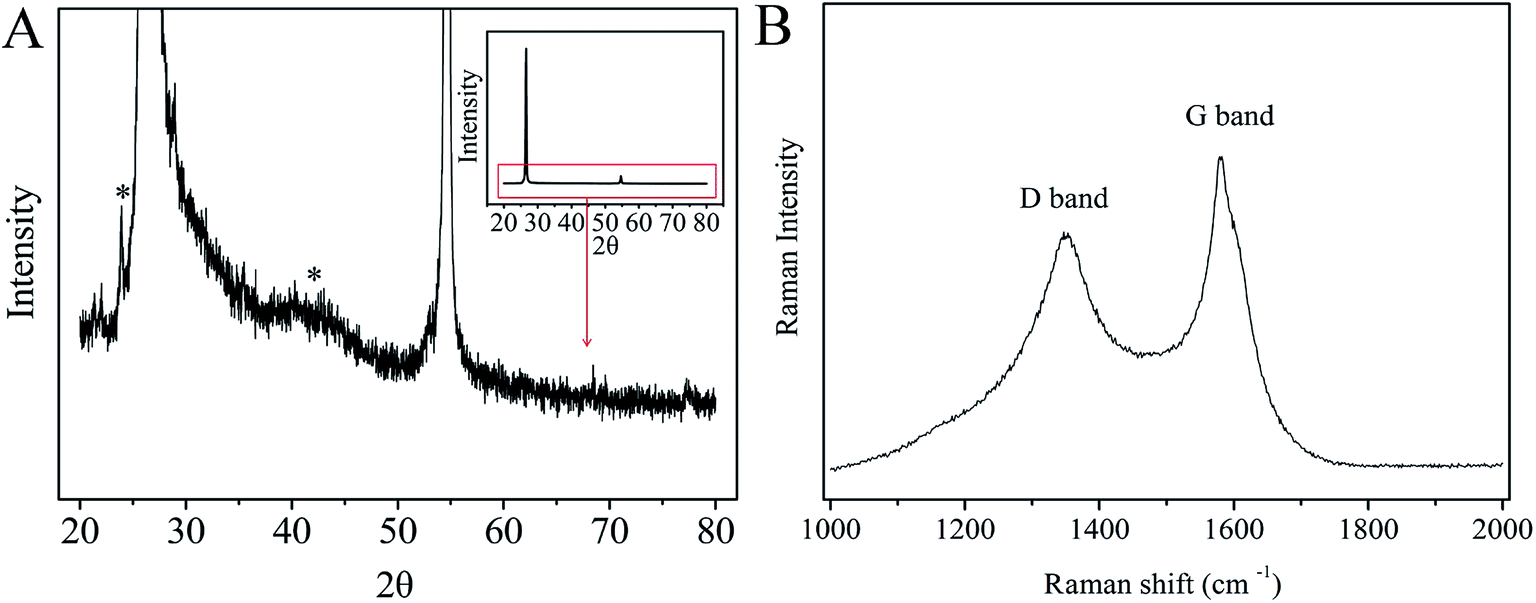
CNTs/CP composite have been characterized by XRD and Raman spectra, as displayed in Fig. 2. The peaks at around 26.5° and 55° are the characteristic (002) and (211) diffraction peaks of carbon paper which is made from expanded graphite, respectively.38–40 However, another two weak peaks appeared at around 25° and 43° on the XRD pattern of the CNTs/CP composite, which should be ascribed to the characteristic diffraction peaks of CNTs.41–43 The thickness of CNTs layer is only about 10 μm, therefore, compared with the characteristic peaks of the substrate, the characteristic diffraction peaks of the graphite from CNTs is relatively quite weak. Raman spectra clearly show the D and G bands at 1355 cm−1 and 1582 cm−1, respectively. The D band is induced by defects and disorder sp2 hybridized carbon, whereas the G band is associated with the tangential stretching mode of highly ordered pyrolytic graphite and indicates the presence of crystalline graphitic carbon, as reported by previous works for CNTs.44,45 Although many electrochemical sensors have been developed for sensitive detection of MP and these sensors exhibit better analytical performance, CNTs/CP electrodes have its own advantages: (1) time saving: CNTs/CP can be directly used as the working electrode, which will save the procedure of polishing and modifying electrodes. (2) Avoid dispersion of CNTs:CNTs/CP electrode directly used as the working electrode can elegantly avoid the poor dispersion of CNTs. (3) CNTs/CP can be produced in large-scale. The large-scale fabrication of CNTs/CP can be not only easily and cheaply realized by CVD but also can be easily shapeable, which is very important for the possible application in the future.
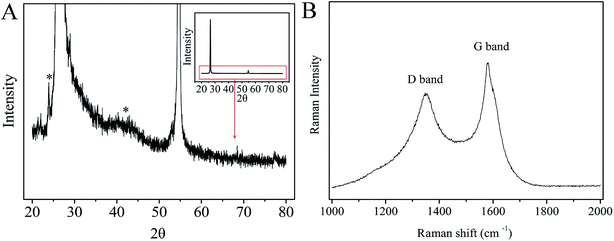 |
| | Fig. 2 (A) The magnified XRD spectra; inset: the original XRD spectra of CNTs/CP composite and (B) Raman spectra of CNTs/CP composite. | |
3.2. Electrochemical behaviour of MP at CNTs/CP electrode
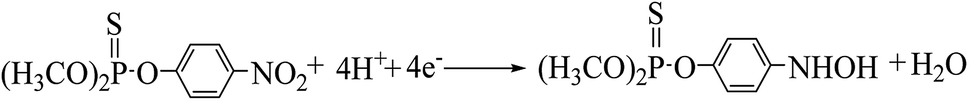
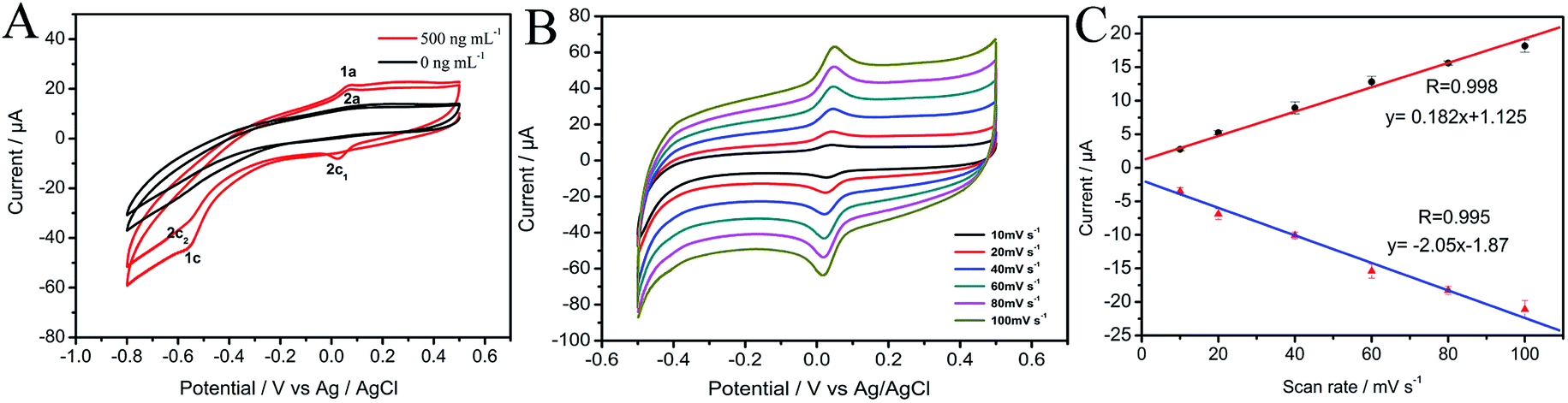
The cyclic voltammogram of 500 ng mL−1 MP at CNTs/CP electrode in 0.2 M acetate buffer shows a pair of well-defined redox peaks (E2a, 0.076 V and E2c1, 0.052) and an irreversible reduction peak (Ec1, −0.54 V) (Fig. 3A). Compared with the electrochemical behavior of CNTs/CP in the absence of MP, it is conferred that the observed oxidation and reduction peaks are produced by electrochemical reaction of MP. The irreversible reduction peak corresponds to the irreversible reduction of the nitro group (–NO2) to hydroxylamine moieties (–NHOH) (reaction (1)), and the reversible redox peaks are attributed to a two-electron-transfer process (reactions (2)) between hydroxylamine moieties (–NHOH), as shown below:| |
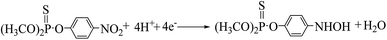 | (1) |
| |
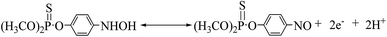 | (2) |
 |
| | Fig. 3 (A) Cyclic voltammograms of 500 ng mL−1 and 0 ng mL−1 MP at CNTs/CP electrode at scan rates of 20 mV s−1; (B) cyclic voltammograms of 500 ng mL−1 MP at CNTs/CP electrode at scan rates of 10, 20, 40, 60, 80, 100 mV s−1 (from inter to outer); (C) plots of the dependence of the anodic and cathodic peak current on the scan rates. | |
The influence of scan rate (ν) on the redox peak current of MP at CNTs/CP electrode was investigated (Fig. 3B and C). Both the anodic and cathodic peak currents of MP are linearly proportional to scan rates in the range of 10–100 mV s−1, confirming that the electrode reaction of MP at CNTs/CP is an adsorption control process. The linear regression equation for anodic and cathodic peak currents are obtained as represented in eqn (3) and (4), respectively.
| | |
Ipa = 0.182ν + 1.125 (R = 0.995)
| (3) |
| | |
Ipc = −0.205ν − 1.87 (R = 0.993)
| (4) |
where
Ipc is the cathodic peak current and
Ipa is the anodic peak current. The
ν stands for the scanning rate in the unit of mV s
−1 and
R is a correlation coefficient.
3.3. Optimization of determination conditions
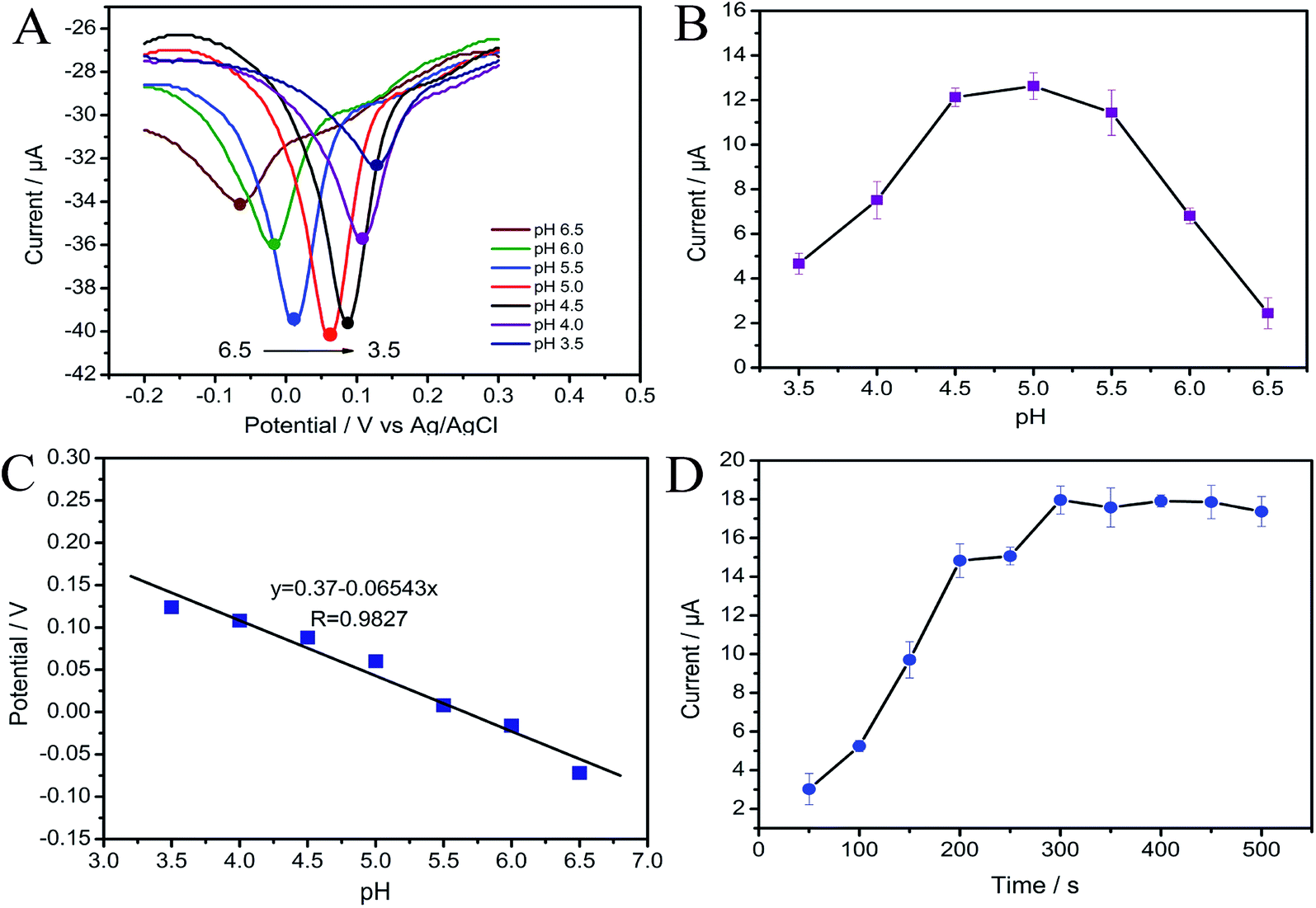
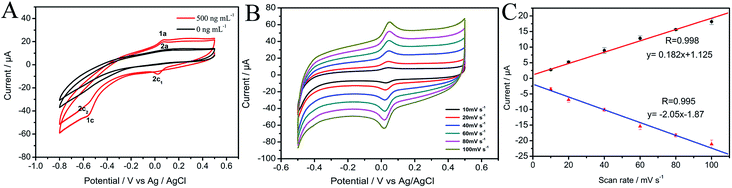
3.3.1 The effect of pH on the response of MP. An important parameter that affected the determination of MP was the pH of the adsorption medium. Redox behavior of MP involves the transfer of electrons and protons, so pH has a great relevance with the buffer solution. Fig. 4A–C exhibit the effect of pH on the reduction peak currents and potentials in the range of 3.5 to 6.5. It can be seen that the peak currents increase gradually when the pH ranges from 3.5 to 5.0, and then decrease with further increasing pH (Fig. 4A and B). The loss of signal at partial acidic or neutral environment may be attributed to the effect of H+ or OH− on adsorption. The response of the MP at high pH values decrease may be ascribed to that the concentration of H+ is insufficient to support the conversion of MP in a reversible manner. At low pH, the decrease of peak current may be attributed to that the high concentration of H+ somewhat prevents the reversible conversion between the –NHOH and –NO.46–48 Therefore, pH 5.0 was chosen as the optimum value in the following test.
 |
| | Fig. 4 (A) Effect of pH on the MP sensor; (B) effect of pH on cathodic peak current in 500 ng mL−1 MP; (C) effect of pH on peak potential in 500 ng mL−1 MP; (D) the influence of adsorption time on cathodic peak current of the DPV response at CNTs/CP, the concentration of MP: 1000 ng mL−1. | |
At the same time, the peak potential (Ep) shows a negative shift with increasing pH, as displayed in Fig. 4C. Furthermore, they show a linear relationship as Ep (V) = −0.06543pH + 0.37, with a correlation coefficient of 0.983 and a slope of 65.43 mV pH−1. According to the Nernstian equation: E ∝ 59.16m/n pH (m represents proton-transfer number, n represents electron-transfer number), we can see that the electrochemical reaction involves equal numbers of proton-transfer and electron-transfer.49
3.3.2 Effect of adsorption time. In order to find out the optimal parameters of CNTs/CP electrode in the detection of MP, we also investigated the relationship of cathodic peak current and the adsorption time. As shown in Fig. 4D, there is no linear relationship between the cathodic peak current and the adsorption time. The peak currents increase rapidly with the adsorption time at first. When the adsorption time is above 300 s, the peak current keeps almost unchanged, meaning that the saturated adsorption of MP onto CNTs/CP electrode is achieved. Therefore, 300 s was chosen as the optimal adsorption time for MP detection.
3.4. Analytical performance
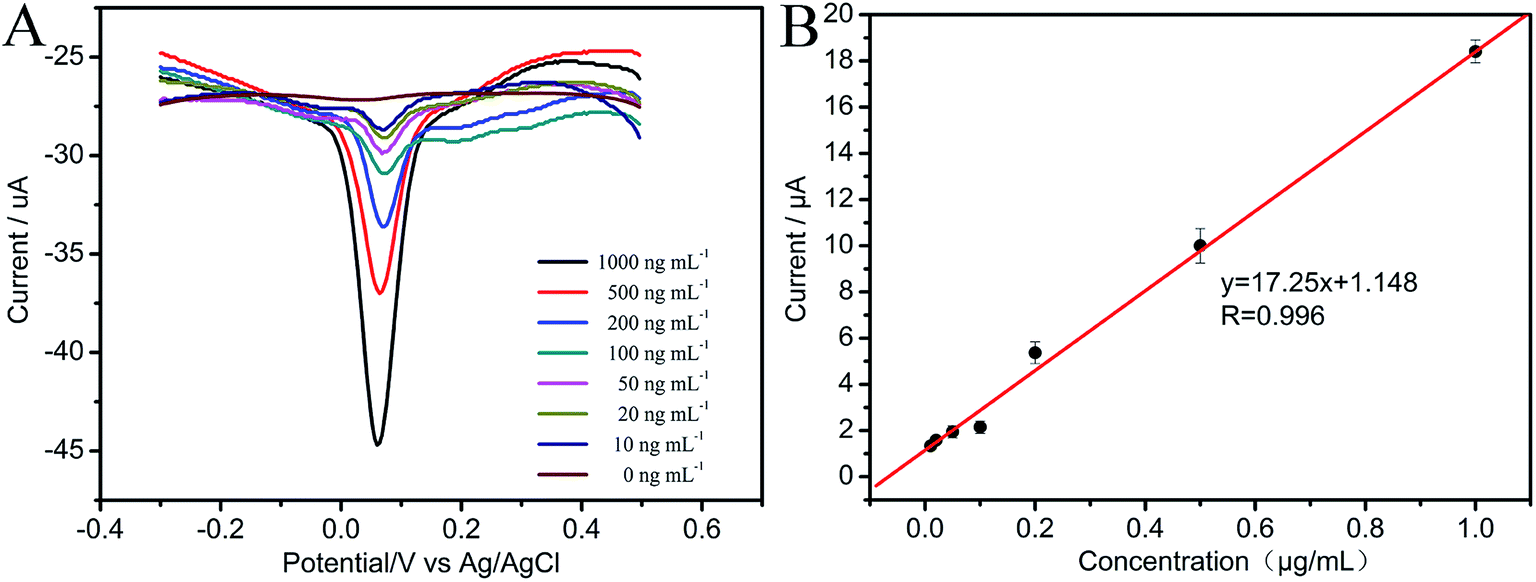
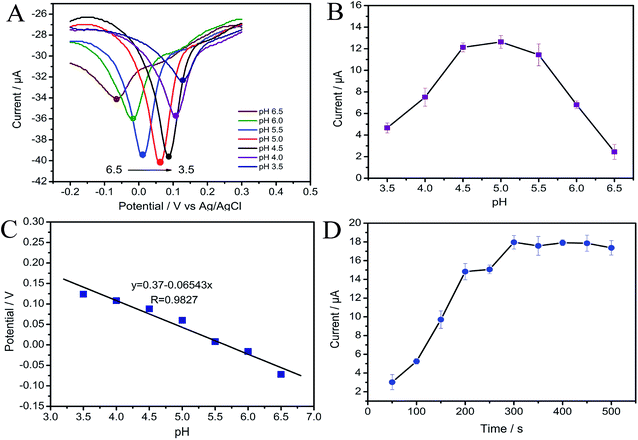
3.4.1 Determination of MP at CNTs/CP electrode. Fig. 5A displays the DPV responses of different concentrations of MP at CNTs/CP electrode under the optimum experimental conditions. Well-defined peaks were observed from Fig. 5A, and linear relationship between the cathodic currents and the MP concentration was obtained. The linear range is from 10 to 1000 ng mL−1 with the linear regression equation being I (μA) = 17.25c (μg mL−1) + 1.148 with a correlation coefficient of 0.996 (Fig. 5B). The detection limit is 3.9 ng mL−1 (S/N = 3), which far exceeded the detection limits of Chinese National Standards for pesticide residues. The detection limit is calculated according to IUPAC recommendations (LOD = 3Sb/b, where Sb is the standard deviation (n = 8) of the blanks, and b is the slope value of the respective calibration graph).49
 |
| | Fig. 5 (A) DPVs of MP in varied concentrations at CNTs/CP; (B) calibration plot of MP. | |
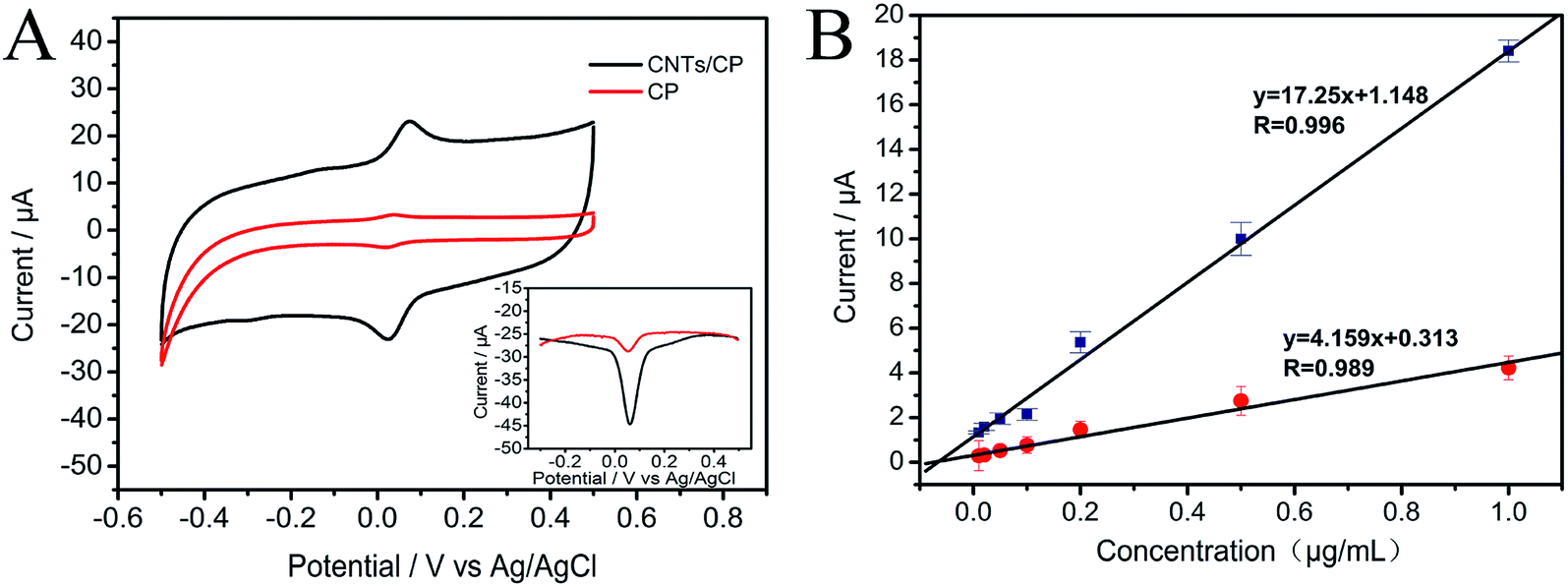
From Fig. 6, it is observed that the slope of the calibration plot of CNTs/CP is much larger than that of the bare CP, confirming the great gain in sensitivity at CNTs/CP electrode. In addition to the sensitivity gain, the improvement of detection limit was also realized by CNTs/CP composite electrode. Besides, the bare CP electrode exhibits a linear relationship in the range of 10–1000 ng mL−1 with a detection limit of 9.4 ng mL−1 at a signal-to-noise ratio of 3. Compared with the bare CP electrode, CNTs/CP composite electrode has a better detection limit (3.9 ng mL−1). This should be ascribed to the huge specific surface area of CNTs, which can facilitate the electrochemical reaction of MP.
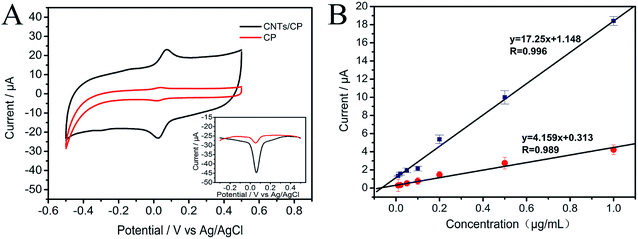 |
| | Fig. 6 (A) Cyclic voltammograms of 1000 ng mL−1 MP at CNTs/CP electrode and CP electrode, respectively. Inset: DPV of 1000 ng mL−1 MP at CNTs/CP electrode and CP electrode (bottom-right) (B) the calibration plots based on the anodic peak currents of different concentrations of MP at CP and CNTs/CP. | |
A comparison analysis on different MP electrochemical sensors reported by previous literatures was shown in Table 1. The CNTs/CP-based electrochemical sensor displayed comparative performance for the trace determination of MP compared with the reported sensors. This is mainly because CNTs/CP electrode combined well with the electrocatalytic activity, fast electron transfer and large surface area of multi-walled carbon nanotubes.12 Some sensors show a little better linear range and detection limit but they normally involved tedious modification processes and noble metal nanoparticles. Furthermore, the detection limit of parathion detected by enzyme inhibition method is 1.0 mg kg−1 according to Chinese National Standards for pesticide residue (GB/T 5009.199-2003). A lower detection limit of 3.9 ng mL−1 is reported in our work. Thus, our simple and inexpensive sensing platform is very promising in the determination of trace MP.
Table 1 Comparison of the proposed electrode with other reported electrodes for the detection of MP
| Electrode |
Technique |
Detection limit (ng mL−1) |
Linear range (ng mL−1) |
Reference |
| Pd–MWCNTs/GCE |
DPV |
50 |
100–14000 |
12 |
| Au–TiO2–Chit/GCE |
DPV |
0.5 |
1–7000 |
13 |
| ZrO2/Au |
SWV |
3 |
5–100 |
3 |
| GR–CNTs–CS/GCE |
SWV |
0.5 |
2–5000 |
48 |
| MWCNT–Chit/GCE |
SWV |
5 |
50–2000 |
50 |
| GR–CS/GCE |
SWV |
0.8 |
4–400 |
51 |
| CNTs/CP |
DPV |
3.8 |
10–1000 |
This work |
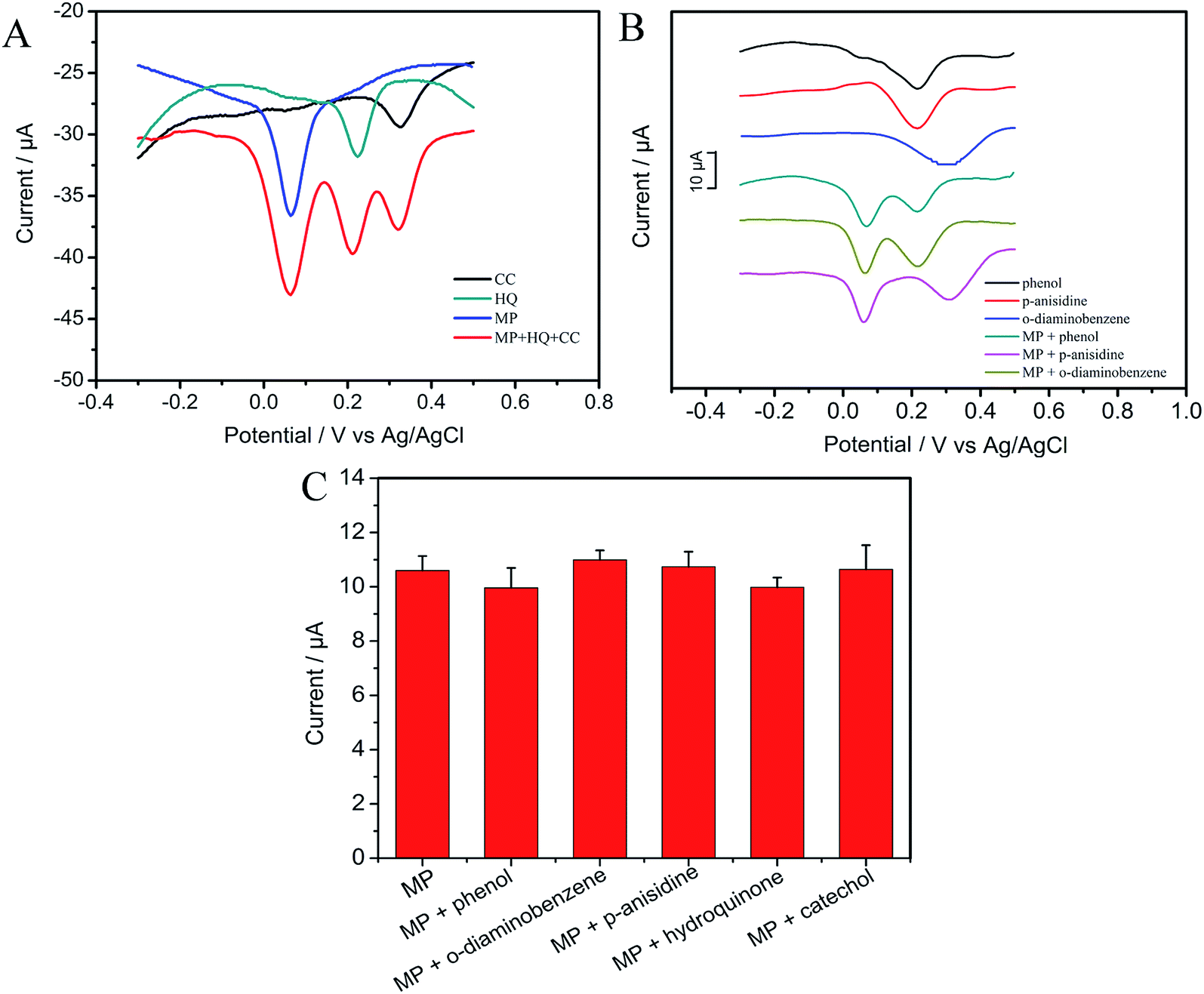
3.4.2 Anti-interference property and stability. To inspect the anti-interference of this self-assembled electrode, five different kinds of common aromatic compounds (catechol, hydroquinone, phenol, p-anisidine and o-diaminobenzene) were used to carry out the interference test at low concentrations (Fig. 7A and B). From the Fig. 7A and B, we can see that compared with the reduction peak current of interferences, the current signals of catechol, hydroquinone, phenol, p-anisidine and o-diaminobenzene without MP almost don't change, indicating that there is no interference between the detection of MP and that of these interferents at CNTs/CP electrode. As illustrated in Fig. 7C, reduction peak current of MP almost does not change in the presence of these five interferents, indicating that these compounds show no interference in the detection of MP at CNTs/CP electrode. The results are consistent with previous works.3,14,46,49 The possible reason is that different compounds have different redox potentials and different adsorption sites on the surface of CP/CNTs, while the adsorption ability of MP may be more competitive.
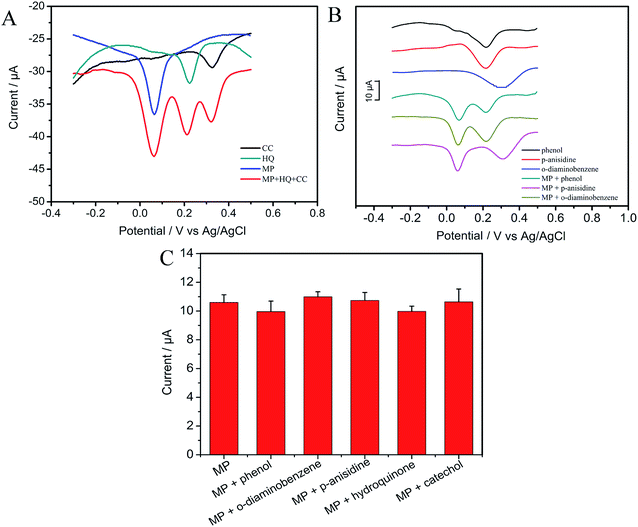 |
| | Fig. 7 DPVs of 500 ng mL−1 MP at the CNTs/CP with the presence of 500 ng mL−1 (A) hydroquinone, catechol, (B) DPVs of 500 ng mL−1 phenol, p-anisidine and o-diaminobenzene in presence or absence of 500 ng mL−1 MP; (C) column diagram of peak current of 500 ng mL−1 MP by DPV in the absence and presence of 500 ng mL−1 phenol, o-diaminobenzene, p-anisidine, catechol and hydroquinone. | |
After the usage of CNTs/CP electrode, the electrode should be immersed in ethanol for 5–10 min to wash the residues. The stability of CNTs/CP electrode can be maintained by being stored at room temperature in a dry condition. No obvious decrease in the response of MP was observed after a 30 day storage period. All these experiments exhibit excellent stability and good anti-interference capability of CNTs/CP electrode.
3.4.3 Application. In order to evaluate the practical feasibility of the electrochemical sensor, the determination of MP in kiwi samples was performed. The samples were in triplicate for detecting. Different concentrations (0, 1.0, 2.0, 4.0, 6.0, 8.0 and 10.0) of standard MP solutions were added to the sample solutions as shown in the Table 2. There is no MP was detected in blank samples. The relative standard deviation (RSD, %) of the repeated measurements was calculated to determine the precision of the method. The recovery was often calculated to evaluate the reproducibility. The RSDs and recoveries of the measured samples were 3.74–5.05% and 99.67–108%, respectively (Table 2), which meets the requirement well for detecting pesticide residues. For future application, according to previous reports, to overcome from this “argon gas dependency”, the testing buffer can be deoxidized by deaerator or catalysts including transition metal oxides, noble metal catalyst and activated carbon for on-site testing.52
Table 2 The results of MP detected by the developed sensor in kiwi samples (n = 3)
| Spiked concentration (μg g−1) |
Detected concentration by sensor (μg g−1) |
RSD by sensor (%) |
Recovery by sensor (%) |
| 0 |
ND |
— |
— |
| 2.0 |
2.13 |
3.74 |
106.50 |
| 4.0 |
4.21 |
4.40 |
105.25 |
| 6.0 |
5.98 |
3.95 |
99.67 |
| 8.0 |
8.64 |
4.99 |
108.00 |
| 10.0 |
10.60 |
5.05 |
106.00 |
4. Conclusion
In summary, an easy-to-fabricate, reusable, low production cost and even disposable CNTs/CP electrode was constructed and used for sensitive determination of MP in the range of 10 ng mL−1 to 1000 ng mL−1 with a low detection limit of 3.9 ng mL−1. Moreover, accumulation time, pH values and interferences of other common aromatic compounds during MP detection have been examined. Finally, sensitive and accurate detection of methyl parathion has been realized in fruit samples (kiwi samples) at the CNTs/CP electrode which demonstrates the applicability of this approach. Importantly, the prepared CNTs/CP could be directly used as working electrode without the polishing procedure that is a necessary procedure for glassy carbon electrode and gold electrode, which can simplify operating steps of MP detection. In a word, this developed electrode construction provided an economic, convenient and practical platform for the detection of MP.
Acknowledgements
This work is financially supported by National Science and Technology Pillar Program (2012BAK17B06), National Natural Science Foundation of China (Grant No. 21103214, 31101274), the Shaanxi Provincial Research Fund (2014K13-10, 2012KJXX-17), Open Fund of State Key Laboratory of Electroanalytical Chemistry (SKLEAC201301), and Program for New Century Excellent Talents in University (NCET-13-0483), Fundamental Research Funds for the Northwest A&F University of China (2014YB093), and the Research Program of Qingdao Municipal Science and Technology Commission (No. 12-1-4-9-(4)-jch).
References
- T. Kang, F. Wang, L. Lu, Y. Zhang and T. Liu, Sens. Actuators, B, 2010, 145, 104–109 CrossRef CAS.
- T. Zhang, L. Zeng, L. Han, T. Li, C. Zheng, M. Wei and A. Liu, Anal. Chim. Acta, 2014, 822, 23–29 CrossRef CAS PubMed.
- G. Liu and Y. Lin, Anal. Chem., 2005, 77, 5894–5901 CrossRef CAS PubMed.
- F. Kayhan, G. Kaymak and N. D. Yön, J. Marmara Univ. Inst. Health Sci., 2013, 4, 167 Search PubMed.
- C. G. Zambonin, M. Quinto, N. De Vietro and F. Palmisano, Food Chem., 2004, 86, 269–274 CrossRef CAS.
- A. Bouaid, L. Ramos, M. J. Gonzalez, P. Fernández and C. Cámara, J. Chromatogr. A, 2004, 939, 13–21 CrossRef.
- F. Ahmadi, Y. Assadi, S. M. Hosseini and M. Rezaee, J. Chromatogr. A, 2006, 1101, 307–312 CrossRef CAS PubMed.
- K. Seebunrueng, Y. Santaladchaiyakit and S. Srijaranai, Chemosphere, 2014, 103, 51–58 CrossRef CAS PubMed.
- J. Wang and M. P. Chatrathi, Anal. Chem., 2001, 73, 1804–1808 CrossRef CAS PubMed.
- W. Wu, Y. Wu, M. Zheng, L. Yang, X. Wu, X. Lin and H. Zeng, Analyst, 2010, 135, 2150–2156 RSC.
- G. Liu and Y. Lin, Electrochem. Commun., 2005, 7, 339–343 CrossRef CAS.
- B. Huang, W. Zhang, C. Chen and Y. Yu, Microchim. Acta, 2010, 171, 57–62 CrossRef CAS.
- Y. Qu, H. Min, Y. Wei, F. Xiao, G. Shi, X. Li and L. Jin, Talanta, 2008, 76, 758–762 CrossRef CAS PubMed.
- J. Gong, X. Miao, T. Zhou and L. Zhang, Talanta, 2011, 85, 1344–1349 CrossRef CAS PubMed.
- M. Musameh, M. R. Notivoli, M. Hickey, C. P. Huynh, S. C. Hawkins, J. M. Yousef and I. L. Kyratzis, Electrochim. Acta, 2013, 101, 209–215 CrossRef CAS.
- W. Zhang, A. M. Asiri, D. Liu, D. Du and Y. Lin, TrAC, Trends Anal. Chem., 2014, 54, 1–10 CrossRef CAS.
- Y. Yao, L. Zhang, J. Xu, X. Wang, X. Duan and Y. Wen, J. Electroanal. Chem., 2014, 713, 1–8 CrossRef CAS.
- J. Ma and W. Zhang, Microchim. Acta, 2011, 175, 309–314 CrossRef CAS.
- B. Sanghavi, G. Hirsch, S. P. Karna and A. K. Srivastava, Anal. Chim. Acta, 2012, 735, 37–45 CrossRef CAS PubMed.
- X. Wang, S. Wang and Z. Cai, TrAC, Trends Anal. Chem., 2013, 52, 170–185 CrossRef CAS.
- F. Yan, Y. He, L. Ding and B. Su, Anal. Chem., 2015, 87, 4436–4441 CrossRef CAS PubMed.
- W. Tang, J. Zhou, Q. Yang, Q. Zheng and J. Wu, Anal. Methods, 2014, 6, 5886–5890 RSC.
- X. Xue, Q. Wei, D. Wu, H. Li, Y. Zhang and R. Feng, et al., Electrochim. Acta, 2014, 116, 366–371 CrossRef CAS.
- B. Wu, L. Hou, M. Du, T. Zhang, Z. Wang and Z. Xue, et al., RSC Adv., 2014, 4, 53701–53710 RSC.
- J. Dong, X. Wang, F. Qiao, P. Liu and S. Ai, Sens. Actuators, B, 2013, 186, 774–780 CrossRef CAS.
- X. Fu, J. Zhang, Y. Tao, J. Wu, C. Xie and L. Kong, Electrochim. Acta, 2015, 153, 12–18 CrossRef CAS.
- H. Liang, X. Miao and J. Gong, Electrochem. Commun., 2012, 20, 149–152 CrossRef CAS.
- M. Xu, J. Zhu, H. Su, J. Dong, S. Ai and R. Li, J. Appl. Electrochem., 2012, 42, 509–516 CrossRef CAS.
- Y. Li, M. Xu, P. Li, J. Dong and S. Ai, Anal. Methods, 2014, 6, 2157–2162 RSC.
- U. Guth, W. Vonau and J. Zosel, Meas. Sci. Technol., 2009, 20, 042002 CrossRef.
- Y. Zhang, T. Kang, Y. Wan and S. Chen, Microchim. Acta, 2009, 165, 307–311 CrossRef CAS.
- L. Dai, D. Chang, J. Baek and W. Lu, Small, 2012, 8, 1130–1166 CrossRef CAS PubMed.
- C. H. Wang, H. Y. Du, Y. T. Tsai, C. P. Chen, C. J. Huang, L. C. Chen, K. H. Chen and H. C. Shih, J. Power Sources, 2007, 171, 55–62 CrossRef CAS.
- I. Lahiri, S. W. Oh, J. Y. Hwang, S. Cho, Y. K. Sun, R. Banerjee and W. Choi, ACS Nano, 2010, 4, 3440–3446 CrossRef CAS PubMed.
- D. Pan, M. Ombaba, Z. Y. Zhou, Y. Liu, S. W. Chen and J. Lu, ACS Nano, 2012, 6, 10720–10726 CrossRef CAS PubMed.
- H. C. Hsu, C. H. Wang, S. K. Nataraj, H. C. Huang, H. Y. Du, S. T. Chang, L. C. Chen and K. H. Chen, Diamond Relat. Mater., 2012, 25, 176–179 CrossRef CAS.
- C. Wang, X. He, L. Tong, Q. Peng, R. Wang, Y. Li and Y. Li, Composites, Part A, 2013, 50, 1–10 CrossRef CAS.
- S. Gantayat, G. Prusty, D. R. Rout and S. K. Swain, New Res. Carbon Mater., 2015, 30, 432–437 CrossRef.
- P. Sobolciak, M. Mrlík, M. A. AlMaadeed and I. Krupa, Thermochim. Acta, 2015, 617, 111–119 CrossRef CAS.
- M. Chen, B. Lou, Z. Ni and B. Xu, Electrochim. Acta, 2015, 165, 105–109 CrossRef CAS.
- B. Gao, C. Peng, G. Chen and G. Lipuma, Appl. Catal., B, 2008, 85, 17–23 CrossRef CAS.
- S. Wang and S. Zhou, J. Hazard. Mater., 2011, 185, 77–85 CrossRef CAS PubMed.
- C.-M. Chen, Y.-M. Dai, J. G. Huang and J.-M. Jehng, Carbon, 2006, 44, 1808–1820 CrossRef CAS.
- C. R. C. Chitturi Venkateswara Rao and Y. Ishikawa, J. Phys. Chem.
Lett., 2010, 1, 2622–2627 CrossRef.
- O. Valentino, M. Sarno, N. G. Rainone, M. R. Nobile, P. Ciambelli and H. C. Neitzert, et al., Phys. E, 2008, 40, 2440–2445 CrossRef CAS.
- L. Zhao, F. Zhao and B. Zeng, Sens. Actuators, B, 2013, 176, 818–824 CrossRef CAS.
- D. Pan, S. Ma, X. Bo and L. Guo, Microchim. Acta, 2011, 173, 215–221 CrossRef CAS.
- Y. Liu, S. Yang and W. Niu, Colloids Surf., B, 2013, 108, 266–270 CrossRef CAS PubMed.
- Q. Sheng, R. Liu, S. Zhang and J. Zheng, Biosens. Bioelectron., 2014, 51, 191–194 CrossRef CAS PubMed.
- D. Du, M. Wang, J. Zhang, J. Cai, H. Tu and A. Zhang, Electrochem. Commun., 2008, 10, 85–89 CrossRef CAS.
- S. Yang, S. Luo, C. Liu and W. Wei, Colloids Surf., B, 2012, 96, 75–79 CrossRef CAS PubMed.
- G. J. S. Dawes, E. L. Scott, J. Le Nôtre, J. P. M. Sanders and J. H. Bitter, Green Chem., 2015, 17, 3231–3250 RSC.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.