
Diterpenoids from the shed trunk barks of the endangered plant Pinus dabeshanensis and their PTP1B inhibitory effects†
Chang-Ling Hu‡
a,
Juan Xiong‡a,
Li-Xin Gaob,
Jia Lib,
Huaqiang Zengc,
Yike Zoud and
Jin-Feng Hu*a
aDepartment of Natural Products Chemistry, School of Pharmacy, Fudan University, No. 826 Zhangheng Road, Shanghai 201203, PR China. E-mail: jfhu@fudan.edu.cn; Fax: +86-21-51980172; Tel: +86-21-51980172
bState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, No. 555 Zuchongzhi Road, Shanghai 201203, PR China
cInstitute of Bioengineering and Nanotechnology, 31 Biopolis Way, The Nanos 138669, Singapore
dDepartment of Chemistry, University of Pennsylvania, 231 South 34 Street, Philadelphia, Pennsylvania 19104-6323, USA
First published on 16th June 2016
Abstract
Rare and endangered plants have proven to be better sources for drug discovery than other botanic sources. Pinus dabeshanensis is an endangered plant listed in the China Plant Red Data Book, and has never been phytochemically investigated. In the present study, 11 new (dabeshanensins A–K, 1–11, resp.) and 28 related known naturally occurring diterpenoids were isolated from the shed trunk bark of this plant. The new structures were established by extensive spectroscopic methods and molecular modeling. Among them, dabeshanensin C (3) has an unprecedented bicyclo[3.3.1]nonane ring system (rings B and C) by oxidative cleavage of the C-6/C-7 bond followed by the formation of a new C–C bond between C-6 and C-12. The plausible biosynthetic pathway to 3 is briefly discussed. Dabeshanensins A (1) and J (10), 12-hydroxydehydroabietic acid (33), abieta-8,11,13,15-tetraen-18-oic acid (35), and 15-hydroxy-7-oxo-8,11,13-abietatrien-18-oic acid (37) showed significant inhibitory effects against the human protein tyrosine phosphatase 1B (PTP1B) enzyme, a key target for the treatment of type-II diabetes and obesity, with IC50 values ranging from 5.4 to 50.9 μM.
Introduction
It has been estimated by the International Union for Conservation of Nature (IUCN) that as many as half of the world's plant species may qualify as threatened with extinction.1 The IUCN's latest Red List revealed the ongoing threats to global plant biodiversity and assessed about 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 endangered or threatened plant species (http://www.iucnredlist.org). The situation is much more serious in China, which ranks third in the world for plant diversity (after Brazil and Colombia).2 In 1992, the first volume of the China Plant Red Data Book (CPRDB) was published, which described 388 endangered taxa warranting protection. Of these, 121 were listed as ‘endangered’, 110 as ‘rare’, and 157 as ‘vulnerable’ (or ‘threatened’).3 The second volume of the CPRDB is now being written, and will contain 640 additional species.2 Meanwhile, plant-derived natural products have continued to play a significant role in the drug discovery and development process.4 The opportunity for identifying new chemical entities (NCEs) for emerging diseases depends on a diversity of drug-producing species. The decline in plant species numbers will continue to exacerbate complication in the discovery of new naturally derived drugs.1a Recently, several statistical surveys unveiled that the rare and endangered plants have proven to be better sources for drug discovery than other botanic sources.5 Therefore, there is a tremendous need to prioritize protection and utilization of these endangered plants at risk and facing extinction.5a
000 endangered or threatened plant species (http://www.iucnredlist.org). The situation is much more serious in China, which ranks third in the world for plant diversity (after Brazil and Colombia).2 In 1992, the first volume of the China Plant Red Data Book (CPRDB) was published, which described 388 endangered taxa warranting protection. Of these, 121 were listed as ‘endangered’, 110 as ‘rare’, and 157 as ‘vulnerable’ (or ‘threatened’).3 The second volume of the CPRDB is now being written, and will contain 640 additional species.2 Meanwhile, plant-derived natural products have continued to play a significant role in the drug discovery and development process.4 The opportunity for identifying new chemical entities (NCEs) for emerging diseases depends on a diversity of drug-producing species. The decline in plant species numbers will continue to exacerbate complication in the discovery of new naturally derived drugs.1a Recently, several statistical surveys unveiled that the rare and endangered plants have proven to be better sources for drug discovery than other botanic sources.5 Therefore, there is a tremendous need to prioritize protection and utilization of these endangered plants at risk and facing extinction.5a
The Pinaceae family ranked among the top 20 privileged drug-prolific families that produced high numbers of approved drugs.5b This family comprises a total of ca. 230 species in 10 genera (e.g., Pinus, Abies, Pseudolarix, Cathaya and Larix).6 As the largest genus of the Pinaceae family, Pinus contains more than 110 species, of which 41 species and 12 varieties are distributed in China.6 Some species of this genus are used in traditional Chinese medicines (TCMs) to treat a variety of ailments such as skin diseases, burn and scald wounds, insomnia, tracheitis, and arthritis.7 Previous studies have revealed that pimarane-, labdane- and abietane-type diterpenoids are the main secondary metabolites of Pinus species with broad bioactivities, such as anti-inflammatory and anti-microbial properties.8 The endangered Pinus plant P. dabeshanensis Cheng et Law is a tall evergreen tree found only in the Dabie (Ta-pieh) Mountains of Central China.6 It is listed in the CPRDB and has been subsequently categorized at the ‘second-grade’ for national protection since 1999.3b In a preceding work,9 a critically endangered conifer, Abies beshanzuensis, has been phytochemically investigated and a few rare enone-containing sesquiterpenoids were obtained with considerable inhibition of the human protein tyrosine phosphatase 1B (PTP1B) enzyme. PTP1B is a negative regulator of the insulin receptor and thus is regarded as a promising drug target for the treatment of type-II diabetes and obesity.10 The same bioassay9,11 revealed that the EtOAc-soluble fraction of the MeOH extract of the shed trunk barks of P. dabeshanensis could significantly inhibit PTP1B. An investigation of this fraction led to the isolation and characterization of 11 new and 28 known diterpenoids with diverse structures (1–39). Herein we report their isolation, structure elucidation, and PTP1B inhibitory activities.
Results and discussion
The 90% MeOH extract (ca. 50 g, semi-dry) of the mass-limited (180 g) shed trunk bark of P. dabeshanensis was suspended in H2O and then partitioned successively with petroleum ether, EtOAc, and n-BuOH. The EtOAc-soluble fraction inhibited PTP1B (IC50 7.61 μg mL−1). Subsequent chromatographic separations of this fraction (ca. 40.0 g) employing silica gel, Sephadex LH-20, and semipreparative HPLC afforded 11 new (1–11) and 28 known (12–39) naturally occurring diterpenoids (Fig. 1). By comparison of their spectroscopic data and physicochemical properties with those reported in the literature, the known ones were identified to be pinusolide acid (12),12 pinusolide (13),13 16-hydroxy-labda-8(17),13-dien-15,19-dioic acid butenolide (14),12b 7-oxo-12α,13β-dihydroxyabiet-8(14)-en-18-oic acid (15),14 7-oxo-13β,15-dihydroxyabiet-8(14)-en-18-oic acid (16),15 7-oxo-13a-hydroxyabiet-8(14)-en-18-oic acid (17),16 abiesadine E (18),17 abiesanordine K (19),17 16-nor-15-oxodehydroabietic acid (20),19 17-nor-7,15-dion-8,11,13-abietatrien-18-oic acid (21),18 methyl 13-acetyl-7-oxo-podocarpa-8,11,13-trien-15-oate (22),20 8(14)-podocarpen-13-on-18-oic acid (23),18 abiesanordine E (24),18 8(14)-podocarpen-7,13-dion-18-oic acid (25),21 7-oxo-13-hydroxy-podocarpa-8,11,13-trien-18-oic acid (26),22 12α-hydroxyabietic acid (27),21 7-oxo-13-epi-pimara-8,15-dien-18-oic acid (28),23 piceanolactones A–C (29–31, resp.),24 dehydroabietic acid (32),25 12-hydroxydehydroabietic acid (33),26 15-hydroxydehydroabietic acid (34),27 abieta-8,11,13,15-tetraen-18-oic acid (35),19 7-oxocallitrisic acid (36),17 15-hydroxy-7-oxo-8,11,13-abietatrien-18-oic acid (37),17 methyl 15-hydroxy-7-oxo-dehydroabietate (38),27 and abiesadine O (39),17 respectively. | ||
| Fig. 1 Structures of compounds 1–39. | ||
Compound 1 was found to possess the molecular formula C20H30O3 based on a quasi-molecular ion peak at m/z 341.2095 ([M + Na]+, calcd 341.2093) in its HRESIMS and by its 13C NMR data, implying six degrees of unsaturation. The 1H and 13C NMR data of 1 (Tables 1 and 3), with the aid of an HSQC NMR experiment (see ESI†), displayed signals of three tertiary methyls [δH 0.75 (s, Me-17), 0.90 (s, Me-20), 1.28 (s, Me-19); δC 15.3 (C-20), 17.1 (C-19), 18.1 (C-17)], eight methylenes, four methines [one olefinic at δH 5.34 (br d, J = 5.2 Hz, H-7); δC 121.3 (C-7)], four quaternary carbons (one olefinic at δC 134.7, C-8), and one carboxyl group at δC 184.7 (C-18). The protons resonating at δH 2.72 (1H, dd, J = 3.6, 3.2 Hz, H-15) and 2.64 (2H, br d, J = 3.5 Hz, H2-16), and carbons at δC 60.6 (C-15) and 43.4 (C-16) suggested the presence of a terminal oxirane ring.28 The above spectroscopic data closely resembled those of 15,16-epoxyisopimaric acid (10), which was semi-synthesized by conversion of isopimaric acid with CYP105A1, with the configuration at C-15 left undetermined.29 Obvious differences were found around the 15,16-epoxy ring (Tables 1 and 3), indicating that the two compounds should have a different configuration at C-13 and/or C-15. Further detailed inspection of the COSY and HMBC spectra of 1 confirmed that it has the same 2D structure as 10 (Fig. 2).
| No. | 1a | 2a | 3a | 4a | 5b |
|---|---|---|---|---|---|
| a Recorded in CDCl3.b Recorded in CD3OD. | |||||
| 1 | 1.86, m | 2.14, br d (13.2) | 1.41, ddd (12.8, 3.7, 3.7) | 1.80, m | 1.88, br d (13.6) |
| 1.13, m | 1.12, ddd (13.2, 13.2, 4.0) | 0.74, ddd (12.8, 12.8, 4.0) | 1.11, m | 1.20, m | |
| 2 | 1.56, m, 2H | 1.93, m | 1.58, m | 1.83, m | 1.62, m, 2H |
| 1.79, m | 1.26, m | 1.52, m | |||
| 3 | 1.77, m | 2.08, br d (13.2) | 1.75, ddd (13.0, 3.4, 3.0) | 2.20, m | 1.88, br d (13.6) |
| 1.68, m | 1.48, ddd (13.2, 13.2, 4.0) | 1.28, m | 1.22, m | 1.64, m | |
| 5 | 1.76, m | 1.39, d (12.0) | 1.86, dd, overlapped | 2.46, dd (13.0, 4.0) | |
| 6 | 1.99, m | 4.49, dd (12.0, 4.8) | 2.18, m | 2.43, dd (17.0, 13.0) | |
| 1.71, m | 2.12, m | 2.39, dd (17.0, 4.0) | |||
| 7 | 5.34, br d (5.2) | 9.50, s | 4.45, br s | ||
| 9 | 1.96, m | 2.68, dd (2.6, 2.6) | 2.20, m | 2.30, m | |
| 11 | 1.58, m | 2.40, ddd (17.5, 6.8, 6.0) | 1.99, m | 1.78, m | 1.72, m |
| 1.36, m | 2.30, m | 1.55, m | 1.61, m | 1.32, m | |
| 12 | 1.45, m | 3.52, m | 2.42, m | 3.77, dd (11.6, 3.6) | |
| 1.39, m | 1.60, m, 2H | 2.21, m | |||
| 14 | 1.94, d (13.6) | 2.56, d (17.2) | 7.57, s | 7.15, br s | 6.72, br d (2.8) |
| 1.85, d (13.6) | 2.27, d (17.2) | ||||
| 15 | 2.72, dd (3.6, 3.2) | 5.76, dd (17.6, 10.8) | 4.80, d (1.2), 2H | 2.17, m | |
| 16 | 2.64, br d (3.5), 2H | 4.94, dd (10.8, 1.2) | 2.00, s | 1.06, d (6.8) | |
| 4.90, dd (17.6, 1.2) | |||||
| 17 | 0.75, s | 1.04, s | 2.08, s | 5.14, br s | 0.86, d (6.8) |
| 4.75, br s | |||||
| 19 | 1.28, s | 1.59, s | 1.17, s | 1.25, s | 1.27, s |
| 20 | 0.90, s | 1.39, s | 1.21, s | 0.60, s | 0.95, s |
 | ||
| Fig. 2 Observed key 1H–1H COSY and HMBC correlations of 1–4, 7 and 8. | ||
The relative stereochemistries of the epoxide groups in 1 and 10 were then determined through the NOESY experiment (Fig. 3) and corroborated by ab initio calculations.30 Similar to 10, clear NOE correlations of Me-19/Me-20, Me-20/H-11β, H-11β/Me-17 and H-5/H-9 were also observed for compound 1. This indicated that Me-17, Me-19 and Me-20 were all β-oriented, and consequently compounds 1 and 10 should be a pair of C-15 epimers. Moreover, the NOE association between Me-17 and H-15 was clearly observed for both 1 and 10, whereas an additional NOE correlation of Me-17/H2-16 was only observable for compound 1 (Fig. 3). Molecular modeling was then incorporated to support the NOE-based relative stereochemistry of the 15,16-epoxy ring. The C-15 epimeric structures [(15R*)-1 and (15S*)-1] were optimized at the level of B3LYP/6-31G(d,p) in CHCl3 and the energies were calculated at the level of B3LYP/6-311+G(2d,p) to calculate the possible low-energy conformers. The 1H–1H interproton distances between H-15 and Me-17, and between H2-16 and Me-17 in the stable conformers for (15R*)-1 and (15S*)-1 were thereafter calculated. We should bear in mind that an NOE correlation could be observed when the Boltzmann-averaged interproton distance is generally less than or approximately 3 Å through space.30,31 As for (15R*)-1, the calculated interproton distances between H-15 and Me-17 (2.63 Å), and between H2-16 and Me-17 (2.83 Å) were approximately consistent with the observed NOE correlations for 1. In contrast, the calculated interproton distances of H-15/Me-17 (2.48/3.08 Å), and H2-16/Me-17 (4.30 Å) for (15S*)-1 matched well with the aforementioned findings in the NOESY spectrum of 10. Thus, the structure of compound 1 was defined as 15(R*),16-epoxy-13-epi-pimara-7-en-18-oic acid (dabeshanensin A), and that of compound 10 as 15(S*),16-epoxy-13-epi-pimara-7-en-18-oic acid.
 | ||
| Fig. 3 Observed key NOE correlations of compounds 1 and 10. | ||
Dabeshanensin B (2) showed an [M + Na]+ ion peak at m/z 335.1629 in its HRESIMS, which in conjunction with the 13C NMR data indicated a molecular formula of C20H24O3, requiring nine degrees of unsaturation. The absorption bands at 1807 and 1675 cm−1 in the IR spectrum of 2 gave hints of the presence of β,γ-unsaturated γ-lactone and conjugated carbonyl functionalities.24 The 1H NMR spectrum of 2 displayed signals of three quaternary methyls [δ 1.04 (s, Me-17), 1.59 (s, Me-19), 1.39 (s, Me-20)] and a terminal vinyl group (ABX pattern) [δ 5.76 (dd, J = 17.6, 10.8 Hz, H-15), 4.94 (dd, J = 10.8, 1.2 Hz, H-16a), and 4.90 (dd, J = 17.6, 1.2 Hz, H-16b)] (Table 1). The 13C NMR data (Table 3), showing twenty carbon signals in total, revealed the presence of three double bonds [δ 144.9 (C-5), 142.7 (C-6), 131.2 (C-8), 159.8 (C-9), 146.1 (C-15), 111.4 (C-16)] and two carbonyl groups [δ 174.7 (C-7), 179.8 (C-18)]. The above spectroscopic data were similar to those of 7-oxo-13-epi-pimara-8,15-dien-18-oic acid (28), a known pimarane-type diterpenoid previously identified from the leaves of Juniperus communis.23 The presence of an additional persubstituted double bond in 2, together with the difference observed for the chemical shift of the C-7 ketone (2: δ 174.7, 28: δ 199.6), hinted the addition of a double bond between C-5 and C-6, conjugated to the C-7 ketone. By now, two carbonyl groups, three double bonds and the tricyclic ring for the pimarane scaffold accounted for eight out of the nine degrees of unsaturation in compound 2. The remaining one was then attributed to the formation of a β,γ-unsaturated γ-lactone ring bridging C-6 and C-18, which was supported by the IR absorption maximum at 1807 cm−1.24 The observed NOE correlations of Me-19/Me-20, Me-20/H-14β and H-14β/Me-17 were in good accordance with the proposed structure of 2 (see ESI†). Thus, compound 2 was characterized as 7-oxo-13-epi-pimara-5,8,15-trien-18,6-olide.
The positive mode HRESIMS of dabeshanensin C (3) gave a quasi-molecular ion peak at m/z 337.1789 [M + Na]+; this along with the 13C NMR data indicated a molecular formula of C20H26O3 with eight degrees of unsaturation. The IR absorptions at 1780 and 1671 cm−1 were, respectively, attributed to the γ-lactone and conjugated carbonyl functionalities. In the 1H NMR spectrum, two tertiary [δ 1.17 (s, Me-19), 1.21 (s, Me-20)] and two vinylic [δ 2.00 (s, Me-16), 2.08 (s, Me-17)] methyls, one oxymethine at δ 4.49 (dd, J = 12.0, 4.8 Hz, H-6), one olefinic proton at δ 7.57 (s, H-14), and one formyl proton at δ 9.50 (s, H-7) were observed (Table 1). The 13C NMR spectrum of 3 exhibited twenty carbon resonances, classified by DEPT and HSQC NMR experiments as four methyls, four methylenes, six methines [one oxygenated at δ 78.9 (C-6), one olefinic at δ 148.0 (C-14), and a formyl at δ 193.3 (C-7)], five quaternary carbons [three olefinic at δ 125.8 (C-13), 138.4 (C-8), and 148.9 (C-15)], and one carboxyl group (δ 181.8, C-18) (Table 3). Taking the molecular formula and the above NMR data into consideration, two double bonds and two carbonyl groups consumed four of the eight degrees of unsaturation. Thus, the remaining unsaturation units required that compound 3 possessed a tetracyclic ring system. More detailed information about the 2D structure of 3 came from inspection of its COSY and HMBC data (Fig. 2). Two spin systems of CH2(1)–CH2(2)–CH2(3) and CH(5)–CH(6)–CH(12)–CH2(11)–CH(9) observed in the 1H–1H COSY spectrum, together with the HMBC correlations of Me-20 with C-1/C-5/C-9/C-10, of Me-19 with C-3/C-4/C-5/C-18, and of H-5 and H2-11 with C-6, led to the construction of rings A and B. Meanwhile, the remaining functionalities (a formyl, a trisubstituted and a persubstituted double bond, and two vinyl methyls) could be easily connected as shown in Fig. 2 due to the cross peaks of the formyl proton (H-7) with C-8, of H-14 with C-7/C-15, of Me-16 and Me-17 with C-15/C-13 in the HMBC spectrum. Furthermore, this fragment was fused with the B-ring via the C-8–C-9 and C-13–C-12 bonds to form the third cyclohexene ring; this fusion was supported by HMBC correlations of H-6 with C-13, of H-7 with C-9, and of H-14 with C-9 and C-12. Finally, as one more ring was required in the structure, the existence of the γ-lactone ring was then readily distinguished. In good agreement with this assumption, the IR absorption band associated with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band stretching (1780 cm−1) typical for a γ-lactone ring was observed.32 Thus, the 2D structure of 3 was determined as depicted in Fig. 1.
O band stretching (1780 cm−1) typical for a γ-lactone ring was observed.32 Thus, the 2D structure of 3 was determined as depicted in Fig. 1.
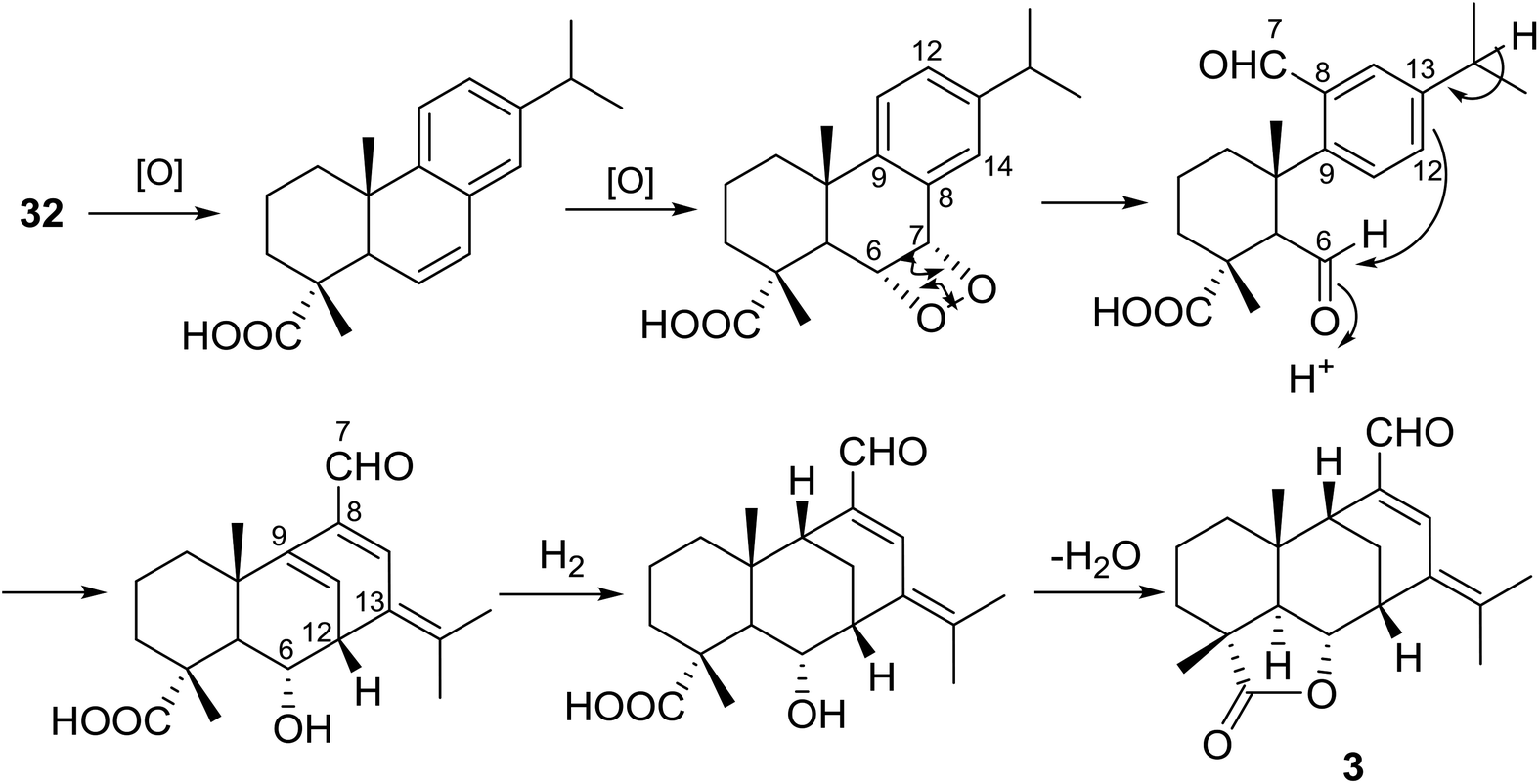
The relative configuration of 3 was assigned by analyses of the proton–proton coupling constants (Table 1) and NOE correlations as shown in Fig. 4. The large coupling constant JH-5,6 (12.0 Hz) and smaller JH-6,12 (4.8 Hz) and JH-9,11 (2.6, 2.6 Hz) implied that H-5 and H-6 were in axial positions, while H-9 and H-12 were equatorial. Clear NOE correlations of Me-20/H-6, Me-19/H-6, H-6/H-7, and Me-20/H-9 indicated that these protons all took the same β-orientations. A plausible biogenetic pathway toward compound 3 was proposed as shown in Scheme 1. Abieta-8,11,13-trien-18-oic acid (32) is considered to be the biosynthetic precursor, which would undergo an enzymatic dehydrogenation followed by an oxidative cleavage of the Δ6,7 double bond to afford the hemiacetal intermediate.33 Subsequent protonation of the C-6 aldehyde group could initiate a concerted cyclization between C-6 and C-12, terminated by loss of a proton at C-15. Following hydrogenation of the Δ9,11 double bond, an intramolecular dehydration between the 18-COOH group and 6-OH could furnish compound 3.
 | ||
| Fig. 4 Observed key NOE correlations of compound 3. | ||
 | ||
| Scheme 1 The proposed biogenetic pathway to compound 3. | ||
Dabeshanensin D (4) has a molecular formula of C20H28O5 as deduced by the HRESIMS (m/z 371.1831 [M + Na]+, calcd 371.1834) and 13C NMR data. A strong IR absorption band at 1721 cm−1 together with the maximum absorption at 217 nm in its UV spectrum denoted the presence of an α,β-unsaturated γ-lactone moiety in the structure of compound 4. This was confirmed by the observed proton signals at δ 4.80 (2H, d, J = 1.2 Hz, H2-15) and 7.15 (1H, br s, H-14) in the 1H NMR spectrum of 1 (Table 1). Meanwhile, signals of two methyl singlets at δ 0.60 (s, Me-20) and 1.25 (s, Me-18), an oxymethine proton at δ 4.45 (br s, H-7), and an exocyclic methylene group at δ 4.75 and 5.14 (both br s, H2-17) were also readily recognized. The 13C NMR spectrum of 4 exhibited twenty carbon resonances (Table 3) comprising two methyls, eight methylenes [one olefinic at δ 109.9 (C-17)], four methines [one oxygenated at δ 73.9 (C-7) and one olefinic at δ 144.6 (C-14)], four quaternary carbons [two olefinic at δ 134.7 (C-13) and 148.2 (C-8)], and two carbonyl groups [one lactone at δ 174.8 (C-16) and one carboxylic at δ 182.5 (C-19)]. The above spectroscopic data showed a great resemblance to those of a known labdane diterpenoid, pinosolide acid (12).12 The only difference between these two compounds is that 4 possesses an additional 7-OH group, which was confirmed by the sequential 1H–1H COSY correlations from H-5 (δ 1.86) to H2-6 (δ 2.12, 2.18), and from H2-6 to H-7 (δ 4.45), together with the key HMBC correlations from H-7 to C-5, C-9 and C-17 (Fig. 2). Moreover, the small coupling constant observed for H-7 (J ≈ 0 Hz) and the NOE correlations of Me-20/H-6β and H-6β/H-7 indicated the β-orientation of H-7 and Me-20, while the correlations of Me-18/H-5 and H-5/H-9 suggested their α-orientations (Fig. 5). Consequently, compound 4 was identified as 7α-hydroxy-15,16-epoxy-16-oxo-labda-8(17),13-dien-19-oic acid.
 | ||
| Fig. 5 Observed key NOE correlations of compound 4. | ||
Dabeshanensin E (5) was assigned the molecular formula C20H30O5 by the HRESIMS (m/z 373.1993 [M + Na]+) and 13C NMR data. The hydroxy, carboxyl, and enone moieties were inferred from the IR absorptions at 3456, 1723, and 1692 cm−1. The 1H NMR spectrum of 5 displayed signals attributed to two singlet [δ 0.95 (s, Me-20), 1.27 (s, Me-19)] and two doublet [δ 0.86 (d, J = 6.8 Hz, Me-17), 1.06 (d, J = 6.8 Hz, Me-16)] methyls, an oxymethine proton [δ 3.77 (dd, J = 11.6, 3.6 Hz, H-12)], and a vinyl proton [δ 6.72 (br d, J = 2.8 Hz, H-14)] (Table 1). Twenty carbon signals were present in the 13C NMR spectrum of 5 (Table 3), including those characteristic of a secondary (δ 69.9, C-12) and a tertiary (δ 74.0, C-13) hydroxy group, an α,β-unsaturated ketone [δ 201.7 (C-7), 137.8 (C-8), 140.6 (C-14)], and a carboxyl group (δ 183.9, C-18). The above spectroscopic data suggested that 5 features an abietane-type diterpenoid, with high similarities to the data of 7-oxo-12α,13β-dihydroxyabiet-8(14)-en-18-oic acid (15), which was isolated from the pine cone of Pinus armandii.14 Detailed comparison of the NMR data between these two compounds suggested that 5 shares the same 2D structure as 15, but requires a different configuration at C-12. This inference was confirmed by the observation of a large coupling constant (11.6 Hz) for H-12, and the NOE correlations of Me-19/Me-20, H-5/H-9, H-9/H-12, H-12/Me-16, and H-12/Me-17 (see ESI†). Thus, the structure of 5 was assigned as 7-oxo-12β,13β-dihydroxy-abieta-8(14)-en-18-oic acid.
Dabeshanensin F (6) has a molecular formula C20H28O4 as determined by HRESIMS (m/z 333.2071 [M + H]+, calcd 333.2060) and 13C NMR data, requiring one more degree of unsaturation than 15. The 1H and 13C NMR data of 6 (Tables 2 and 3) were quite similar to those of 15, except for the presence of an additional Δ13,15 double bond along with the absence of 13-OH in the structure of 6. This was corroborated by the observation of two vinyl methyls [δ 2.00 (s, Me-17) and 2.03 (s, Me-16)] in 6 instead of the corresponding two methyl doublets in 15, and the HMBC correlations of H-14 with C-8/C-9/C-12/C-15, and of Me-16/Me-17 with C-13 and C-15 (see ESI†). The orientations of Me-19, Me-20 and 12-OH in 6 were found to be identical to those in compound 15 according to the J value (close to zero) of H-12 and NOE correlations (see ESI†). Hence, 6 was determined to be 7-oxo-12α-hydroxy-abieta-8(14),13(15)-dien-18-oic acid.
| No. | 6 | 7 | 8 | 9 | 11 |
|---|---|---|---|---|---|
| 1 | 1.80, m | 1.89, br d (13.2) | 2.33, br d (12.4) | 1.78, m | 1.84, m |
| 1.66, m | 1.14, m | 1.48, m | 1.26, m | 1.36, m | |
| 2 | 1.82, m | 1.60, m | 1.79, m, 2H | 1.70, m, 2H | 1.65, m, 2H |
| 1.65, m | 1.49, m | ||||
| 3 | 1.80, m | 1.71, m, 2H | 1.86, m | 1.81, m | 1.75, m |
| 1.64, m | 1.76, m | 1.68, m | 1.66, m | ||
| 5 | 2.44, dd, overlapped | 2.00, br d (11.8) | 2.29, dd (12.0, 2.6) | 2.87, br s | 3.10, s |
| 6 | 2.42, dd, overlapped | 4.35, br d (11.8) | 1.95, m | 6.06, dd (10.0, 2.4) | |
| 2.40, dd, overlapped | 1.90, m | ||||
| 7 | 6.72, br s | 4.95, dd (10.0, 7.6) | 6.29, dd (10.0, 3.0) | 5.77, s | |
| 9 | 2.57, br d (11.2) | 1.96, br d (10.8) | 2.43, ddd (11.6, 3.6, 2.0) | 2.40, m | |
| 11 | 1.35, m | 1.81, m | 7.34, d (8.4) | 2.04, m | 1.92, m |
| 1.24, m | 1.45, m | 1.72, m | 1.42, m | ||
| 12 | 4.86, br s | 3.17, ddd (17.0, 11.0, 5.6) | 7.83, dd (8.4, 2.0) | 2.57, ddd (17.4, 2.7, 2.5) | 2.33, m |
| 2.47, ddd (17.0, 11.0, 5.0) | 2.32, ddd (17.4, 13.0, 4.2) | 2.23, m | |||
| 14 | 7.66, br s | 9.52, s | 8.15, d (2.0) | 5.87, br s | 6.03, s |
| 15 | 2.65, m | 2.38, m | |||
| 16 | 2.03, s | 1.13, d (6.8) | 2.60, s | 1.08, d (6.8) | |
| 17 | 2.00, s | 1.13, d (6.8) | 1.08, d (6.8) | ||
| 19 | 1.28, s | 1.34, s | 1.32, s | 1.26, s | 1.42, s |
| 20 | 0.86, s | 0.87, s | 1.31, s | 0.84, s | 0.95, s |
| No. | 1a | 2a | 3a | 4a | 5b | 6a | 7a | 8a | 9a | 11a |
|---|---|---|---|---|---|---|---|---|---|---|
| a Recorded in CDCl3.b Recorded in CD3OD.c c–hInterchangeable or overlapped within the same superscript in the same column. | ||||||||||
| 1 | 38.8 | 40.4 | 36.0 | 38.8 | 37.5 | 37.7 | 38.3 | 38.3 | 37.2 | 37.8 |
| 2 | 17.9 | 18.6 | 18.9 | 19.8 | 19.1 | 17.7 | 17.3 | 18.2 | 17.6 | 17.6 |
| 3 | 36.9 | 36.5 | 32.7 | 37.8 | 36.7 | 36.8 | 37.5d | 36.2 | 35.8 | 37.4 |
| 4 | 46.3 | 46.9 | 40.5 | 43.8 | 47.1 | 45.9 | 43.8 | 47.0 | 45.6 | 42.9 |
| 5 | 52.0 | 144.9 | 47.9 | 48.4 | 46.0 | 44.0 | 51.5 | 43.1 | 50.6f | 60.2 |
| 6 | 25.2 | 142.7 | 78.9 | 32.0 | 39.9 | 38.1 | 67.3 | 32.5 | 140.0 | 199.2 |
| 7 | 121.3 | 174.7 | 193.3 | 73.9 | 201.7 | 199.7 | 152.2 | 70.3 | 129.6 | 122.1h |
| 8 | 134.7 | 131.2 | 138.4 | 148.2 | 137.8 | 132.6 | 144.1 | 138.1 | 157.9 | 156.6 |
| 9 | 44.9 | 159.8 | 38.2 | 48.8 | 45.4 | 44.2 | 49.0 | 154.3 | 50.7f | 51.2 |
| 10 | 35.0 | 40.9 | 37.6 | 40.4 | 37.4 | 34.7 | 37.5d | 37.6 | 38.1 | 40.0 |
| 11 | 19.4 | 22.4 | 26.7 | 21.8 | 28.2 | 29.4 | 20.8 | 124.7 | 21.2 | 21.3 |
| 12 | 33.3 | 32.7c | 35.4 | 23.8 | 69.9 | 64.5 | 42.3 | 127.4 | 36.5 | 27.6 |
| 13 | 33.9 | 34.4 | 125.8 | 134.7 | 74.0 | 130.2 | 215.3 | 135.2 | 200.9 | 159.9 |
| 14 | 42.0 | 32.9c | 148.0 | 144.6 | 140.6 | 132.1 | 195.4 | 128.1 | 125.4 | 122.1h |
| 15 | 60.6 | 146.1 | 148.9 | 70.2 | 34.3 | 145.7 | 40.7 | 198.1 | 35.7 | |
| 16 | 43.4 | 111.4 | 23.1 | 174.8 | 18.2 | 21.4 | 18.3e | 26.6 | 21.1 | |
| 17 | 18.1 | 25.9 | 21.4 | 109.9 | 18.9 | 20.8 | 18.2e | 20.5 | ||
| 18 | 184.7 | 179.8 | 181.8 | 182.5 | 183.9 | 182.4 | 185.7 | 182.9 | 182.7 | 183.4 |
| 19 | 17.1 | 21.2 | 15.1 | 28.7 | 17.0 | 16.2 | 16.7 | 16.3 | 17.5 | 17.2 |
| 20 | 15.3 | 21.0 | 19.9 | 11.9 | 14.8 | 14.6 | 14.8 | 25.2 | 13.8 | 14.7 |
The molecular formula of dabeshanensin G (7) was established to be C20H30O5 based on 13C NMR data and a quasi-molecular ion at m/z 373.1997 [M + Na]+ in its HRESIMS. The IR absorption bands indicated the presence of hydroxy (3489 cm−1), ketone (1714 cm−1), carboxyl (1699 cm−1) and α,β-unsaturated formyl (1696, 1621 cm−1) groups; the latter was confirmed by the maximum absorption at 242 nm in the UV spectrum of 7. Analysis of its 1H and 13C NMR data (Tables 2 and 3) revealed that compound 7 has four methyls, an α,β-unsaturated aldehyde group [δH 6.72 (br s, H-7), 9.52 (s, H-14); δC 152.2 (C-7), 144.1 (C-8), 195.4 (C-14)], a carboxyl [δC 185.7 (C-18)] and a saturated ketone [δC 215.3 (C-13)] group. These functionalities are characteristic of a 13,14-seco-abietane skeleton similar to 13,14-seco-13,14-dioxoabieta-7-en-18-oic acid, which was previously isolated from the cones of Larix kaempferi.34 The only difference between these two compounds was that 7 has an additional secondary hydroxy group [δH 4.35 (br d, J = 11.8 Hz); δC 67.3], which was concluded to be sited at C-6 based on the observation of a spin system of H(5)–H(6)–H(7) and correlations from H-5 to C-6 in the HMBC NMR spectrum (Fig. 2). The large proton–proton coupling constant (11.8 Hz) between H-5 and H-6, and NOE correlations of Me-19/Me-20, Me-20/H-6 and H-6/Me-19 (see ESI†) unequivocally assigned a β-axial orientation for H-6. Compound 7 was therefore assumed to be 13,14-seco-6α-hydroxy-13,14-dioxo-abieta-7-en-18-oic acid.
Dabeshanensin H (8) exhibited an [M + Na]+ ion peak at m/z 339.1570 in the positive HRESIMS, corresponding to the molecular formula C19H24O4. In accordance with the molecular formula, only nineteen carbon signals (Table 3) were detectable in the 13C NMR spectrum of 8. From inspection of the 1H and 13C NMR spectra, a typical carboxyl group (δC 182.9) at C-4, an ABX system benzene ring [δH 7.34 (d, J = 8.4 Hz, H-11), 7.83 (dd, J = 8.4, 2.0 Hz, H-12), 8.15 (d, J = 2.0 Hz, H-14); δC 138.1 (C-8), 154.3 (C-9), 124.7 (C-11), 127.4 (C-12), 135.2 (C-13), 128.1 (C-14)], a secondary hydroxy group [δH 4.95 (dd, J = 10.0, 7.6 Hz, H-7); δC 70.3 (C-7)], and an acetyl group [δH 2.60 (s, Me-16); δC 198.1 (C-15), 26.6 (C-16)] could be easily recognized. The above data are comparable to those of 17-nor-15-oxo-8,11,13-abietatrien-7α-hydroxy-18-oic acid (19, abiesanordine K).18 The most notable difference was the smaller coupling constants observed for H-7 (dd, J = 4.8, 1.5 Hz) in abiesanordine K than in compound 8 (10.0, 7.6 Hz), indicative of a different configuration at C-7. The 2D structure of 8 was then confirmed by COSY and HMBC data (Fig. 2). The β-orientation of H-7 in 8 was substantiated by the NOESY experiment that showed a cross-peak between H-7 and H-5 (see ESI†). Consequently, compound 8 was elucidated to the C-7 epimer of abiesanordine K.
Dabeshanensin I (9) was found to possess the molecular formula C17H22O3 as deduced by its HRESIMS (m/z 297.1463, [M + Na]+) and 13C NMR data. The 1H NMR spectrum showed signals for two singlet methyl groups [δ 0.84 (s, Me-20), 1.26 (s, Me-19)] and three olefinic protons [δ 5.87 (br s, H-14), 6.06 (dd, J = 10.0, 2.4 Hz, H-7), 6.29 (dd, J = 10.0, 3.0 Hz, H-6)] (Table 2). The 13C NMR data of 9 (Table 3) exhibited seventeen carbon resonances consisting of two methyls, five methylenes, five methines [three olefinic at δ 140.0 (C-6), 129.6 (C-7), 125.4 (C-14)], three quaternary carbons [one olefinic at δ 157.9 (C-8)], and two carbonyl groups [one carboxyl at δ 182.7 (C-18), and one ketone at δ 200.9 (C-13)]. The aforementioned spectroscopic data suggested that 9 is a podocarpene diterpenoid similar to 8(14)-podocarpen-13-on-18-oic acid (23),18 except for the presence of a Δ6,7 double bond. This was verified by the observation of an H(5)-H(6)-H(7) spin system in the 1H–1H COSY spectrum of 9, and HMBC correlations of H-5/C-6, H-6/C-8, H-7/C-9, H-14/C-7 and H-14/C-9 (see ESI†). The relative configuration of 9 was then determined based on the clearly observed NOE correlations of Me-19/Me-20 and H-5/H-9, and the absence of an association between Me-19 and H-5. Thus, compound 9 was characterized as 13-oxo-podocarpa-6,8(14)-dien-18-oic acid.
Compound 11 was assigned the molecular formula C20H28O3 as determined by the HRESIMS data (m/z 317.2110, [M + H]+) and 13C NMR data. The 1H and 13C NMR data (Tables 2 and 3) of 11 revealed the presence of two tertiary methyls [δH 0.95 (s, Me-20), 1.42 (s, Me-19); δC 14.7 (C-20), 17.2 (C-19)], an isopropyl group [δH 1.08 (6H, d, J = 6.8 Hz, Me-16 and Me-17), 2.38 (m, H-15); δC 20.5 (C-17), 21.1 (C-16), 35.7 (C-15)], two trisubstituted double bonds [δH 5.77 (s, H-7), 6.03 (s, H-14); δC 122.1 (C-7), 156.6 (C-8), 159.9 (C-13), 122.1 (C-14)], a typical carboxylic acid (δC 183.4, C-18) at C-4, and a conjugated ketone group (δC 199.2, C-6). These data showed general features very similar to those of abietic acid (= abieta-7,13-dien-18-oic acid),17 except for the existence of an additional ketone group at C-6 in 11, which was corroborated by the HMBC correlations from H-5 (δH 3.10, s) and H-7 (δH 5.77, s) to C-6 (see ESI†). The relative configuration of 11 was found to be the same as in abietic acid by analysis of the NOESY spectrum (see ESI†). Accordingly, the structure of 11 was identified as 6-ketoabietic acid. Actually, this compound has been semi-synthesized as early as in 1950s.35 Herein, compound 11 was reported as a new naturally occurring diterpenoid, and its 1H and 13C NMR data were completely assigned for the first time.
All the isolated compounds (except the mass-limited 3) were evaluated for their inhibitory activity against PTP1B, and the results are presented in Table 4. The known PTP1B inhibitor oleanolic acid (IC50 = 3.3 μM)9,11 was used as a positive control in this assay. Of the 38 compounds tested, abieta-8,11,13,15-tetraen-18-oic acid (35) and 15-hydroxy-7-oxo-abieta-8,11,13-trien-18-oic acid (37) exhibited the most potent inhibitory effects, with IC50 values of 5.4 and 10.3 μM, respectively. 12-Hydroxydehydroabietic acid (33), with an abieta-8,11,13-trien-18-oic acid structure similar to 35 and 37, was less active (IC50 = 50.9 μM). Moreover, the two new 13-epi-pimaranes (1 and 10) featuring an uncommon 15,16-epoxy ring, both showed moderate activities (IC50 = 40.9 and 35.2 μM, resp.). The rest were judged inactive (IC50 > 20 μg mL−1). From the above results, the abietane-type diterpenoids with an aromatic C ring (such as 33, 35 and 37) generally showed higher potencies than others. A plausible explanation is that the aromatic ring might facilitate the hydrophobic interaction with the binding site of the enzyme.10b And, this hydrophobic interaction could be enhanced by introduction of a conjugated ketone group at C-7 (comparing 33 and 37) or a conjugated Δ15,16 double bond (in the case of 35). Moreover, for the abieta-8,11,13-trienes (29–39), the presence of a free 18-COOH group may be also important for the PTP1B inhibition because compounds 29–31 and 38 were all inactive, even though they have a larger conjugated system. The most potent compound, abieta-8,11,13,15-tetraen-18-oic acid (35), was selected herein to investigate the inhibition mode on the activity of PTP1B. As shown in Fig. 6, the Lineweaver–Burk plot results revealed this abietane-type diterpenoid is a competitive inhibitor of PTP1B.
 | ||
| Fig. 6 Lineweaver–Burk plot for PTP1B inhibition of compound 35. The PTP1B inhibition was analyzed in the presence of different sample concentrations (0.0, 2.0, 4.0, 6.0, and 8.0 μg mL−1). | ||
Conclusions
The rare and endangered plants are better sources for providing novel lead compounds.5 There is an urgent need to investigate these fragile plant species endemic to China. Recently, we have launched a new program to systematically identify natural products from plants in the CPRDB.9,11,36 In this study, 39 structurally diverse diterpenoids (including 11 new ones) were obtained from only 180 g (semi-dry) of the shed trunk bark of P. dabeshanensis, which is nationally protected as a second-grade species in China. Five isolates (1, 10, 33, 35, and 37) from this endangered plant were found to show significant PTP1B inhibitory effects. To our knowledge, some naturally occurring ent-pimarane-type and ent-kaurane-type diterpenoids have been previously documented to be potent noncompetitive or mixed-type inhibitors of PTP1B.37 Nevertheless, this is the first report on the PTP1B inhibitory activity of abieta-8,11,13-trien-18-oic acids (e.g., 33, 35, and 37). Further enzyme kinetic analysis of 35 revealed that it is a competitive inhibitor of PTP1B. Our findings may contribute to further exploration of the therapeutic potential of these abietane-type diterpenoids in type-II diabetes and obesity.In many cases, investigation of endangered plants is hampered by the difficulty in collecting the plant materials. Each plant species definitely contains a complex and unique microbiome consisting of untold numbers of bacteria and fungi, which might be adept in synthesizing associated plant metabolites.38 Therefore, studies on the secondary metabolites of endophytes associated with the endangered plants provide an alternative strategy for drug discovery as well as the sustainable use and protection of the fragile plant resources. In this study, 27 endophytes have been successfully isolated from a small amount of the fresh stem bark and twigs of the title plant. Further studies on these endophytes are ongoing in our laboratories.
Experimental section
General experimental procedures
Optical rotations were measured with an Autopol IV-T polarimeter. UV and IR spectra were recorded on a Shimadzu UV-2550 and an Avatar 360 ESP FTIR spectrometer, respectively. NMR spectra were recorded on Bruker Avance III 400 MHz or 600 MHz spectrometers. Chemical shifts are expressed in δ (ppm), and referenced to the residual solvent signals. ESIMS were measured on an Agilent 1100 series mass spectrometer; HRESIMS were measured on a Waters Q-TOF Ultima mass spectrometer and an LCT Premier XE mass spectrometer. Semi-preparative HPLC was performed on a Waters e2695 apparatus equipped with a 2998 photodiode array detector, a 2424 evaporative light-scattering detector (ELSD), a SunFire ODS column (5 μm, 250 × 10 mm; flow rate: 3.0 mL min−1) and a LC-2010A HT SHIMADZU normal-phase chirality column (5 μm, 250 × 4.6 mm, ChiRaLPAK, AD-H; flow rate: 1.0 mL min−1). Column chromatography (CC) was performed using silica gel (200–300 mesh, Kang-Bi-Nuo Silysia Chemical Ltd., Yantai, China), and Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). Silica gel-precoated plates (GF254, 0.25 mm, Kang-Bi-Nuo Silysia Chemical Ltd., Yantai, China) were used for TLC detection. Spots were visualized using UV light (254 and/or 365 nm) and by spraying with 5% (v/v) H2SO4–EtOH followed by heating to 120 °C.Plant material
The shed trunk bark of P. dabeshanensis was collected with the authorized permission in December 2012 from Peach Valley (Tao-Hua-Chong), Yingshan County, Hubei Province of China. The plant was identified by Mr Lian Xu (Forestry Bureau of Yingshan County, Hubei Province). A voucher specimen (No. 20121214) was deposited at the Herbarium of the Department of Natural product chemistry, School of Pharmacy at Fudan University.Extraction and isolation
The air-dried dried trunk bark (180.0 g) was extracted three times with 90% MeOH (5 × 0.5 L) at room temperature. The solvent was removed under reduced pressure to give a brown residue (ca. 50.0 g, semi-dry). The entire crude extract was suspended in water and partitioned with petroleum ether (PE), EtOAc and n-BuOH, successively. The EtOAc fraction (ca. 40.0 g) was subjected to silica gel CC, eluted with a gradient of PE–EtOAc (30:1, 15:1, 5:1, 3:1; each 2 L, v/v) and then EtOAc–MeOH (15:1, 5:1, 3:1; each 2 L, v/v) to give eight fractions (Fr. A– Fr. H). Fr. A (613.6 mg) was chromatographed on silica gel with a gradient of PE–EtOAc (10:1 to 5:1, v/v) and purified on Sephadex LH-20 (MeOH) to afford 22 (1.0 mg) and 36 (280.0 mg). Fr. B (1.9 g) was fractionated by a silica gel column with a gradient elution of PE–EtOAc (10:1, 5:1, 2:1, v/v), to provide three subfractions, B1–B3. Compounds 29 (1.6 mg), 32 (5.0 mg), and 35 (8.0 mg) were purified by semipreparative HPLC [CH3CN–H2O (containing 0.05% TFA, v/v) 73:27, v/v; 35: tR = 21.3 min, 29: tR = 24.3 min, 32: tR = 27.4 min] from subfraction B1. Fr. B2 was subjected to semipreparative HPLC [CH3OH–H2O (containing 0.05% TFA, v/v) 77:23, v/v] to afford 34 (8.0 mg, tR = 13.0 min) and 38 (2.0 mg, tR = 10.4 min). Fr. B3 was also separated by semipreparative HPLC [CH3OH–H2O (containing 0.05% TFA, v/v) 70:30, v/v] to furnish 3 (1.4 mg, tR = 15.8 min), 6 (4.5 mg, tR = 19.4 min) and 28 (5 mg, tR = 22.2 min). Fr. C (1.9 g) was divided into three subfractions (Fr. C1–C3) by silica gel CC with a PE–EtOAc gradient (7:1 to 3:1, v/v). Fr. C1 was further purified by Sephadex LH-20 (MeOH) to obtain 13 (100.2 mg) and 33 (28.0 mg). Fr. C2 was subjected to normal-phase HPLC with a chiral column (n-hexane–propanol 94:6, v/v; flow rate, 1.0 mL min−1) to afford compounds 1 (2.2 mg, tR = 9.5 min) and 10 (2.5 mg, tR = 12.5 min). Compound 27 (30.0 mg) was recrystallized from Fr. D (1.8 g). The filtrate was condensed to give a yellow residue, which was chromatographed over silica gel CC with PE–EtOAc (7:1 to 3:1, v/v) and further purified by Sephadex LH-20 (MeOH) to give 11 (5.0 mg), 12 (260.0 mg) and 18 (8.0 mg).
Fr. E (2.1 g) was divided into three subfractions (Fr. E1–E3) by silica gel CC eluted with PE–EtOAc (3:1, v/v). Fr. E1 was purified by semipreparative HPLC [MeOH–H2O (containing 0.05% TFA, v/v) 75:25, v/v] to furnish 2 (2.0 mg, tR = 12.4 min), 17 (2.4 mg, tR = 10.8 min) and 20 (4.0 mg, tR = 17.0 min). Fr. E2 was subjected to Sephadex LH-20 (MeOH) and then semipreparative HPLC [MeOH–H2O (containing 0.05% TFA, v/v) 65:35, v/v] to yield 9 (2.0 mg, tR = 19.5 min), 14 (8.0 mg, tR = 27.2 min), 21 (1.4 mg, tR = 16.9 min), and 26 (4.0 mg, tR = 15.5 min). Compound 37 (120.0 mg) was obtained from fraction F (1.1 g) by repeated CC on silica gel and SephadexLH-20 (MeOH). Compound 31 (16.6 mg) was obtained from Fr. G (2.2 g) by crystallization from EtOAc, and the filtrate was then applied to silica gel CC using a gradient of PE–EtOAc (5:1, 3:1, 1:1; v/v) to afford three subfractions, G1–G3. Compounds 24 and 25 (2.8 mg) were isolated from Fr. G1 by silica gel CC with PE–EtOAc (3:1, v/v), whereas 8 (2.3 mg), 23 (1.4 mg) and 30 (1.4 mg) were purified from Fr. G2 by semipreparative HPLC [MeOH–H2O (containing 0.05% TFA, v/v) 65:35, v/v; 23: tR = 12.7 min, 30: tR = 16.6 min, 8: tR = 26.7 min]. Compound 19 (4.2 mg, tR = 22.7 min) was obtained from Fr. G3 by semipreparative HPLC [MeOH–H2O (containing 0.05% TFA, v/v) 65:35, v/v]. Fr. H (1.6 g) was fractionated by a silica gel column with a PE–EtOAc gradient (3:1, 1:1, 1:2; v/v) to give three subfractions H1–H3. Compound 39 (15.0 mg) was recrystallized from Fr. H1. Fr. H2 was purified by semipreparative HPLC [MeOH–H2O (containing 0.05% TFA, v/v) 65:35, v/v] to furnish 5 (1.5 mg, tR = 19.7 min), 7 (3.2 mg, tR = 22.8 min), and 15 (3.6 mg, tR = 16.7 min). Compounds 4 (2.6 mg, tR = 15.5 min) and 16 (1.8 mg, tR = 16.9 min) were isolated from Fr. H3 by semipreparative HPLC [CH3CN–H2O (containing 0.05% TFA, v/v) 38:62, v/v].
ε) 217 (0.97) nm; IR (KBr) νmax 2922, 2857, 1698, 1452, 1442, 1386, 1366, 1024, 786 cm−1; 1H and 13C NMR data, see Tables 1 and 3; (+) ESIMS m/z 319 [M + H]+, 341 [M + Na]+; HRESIMS m/z 341.2095 [M + Na]+ (calcd for C20H30O3Na, 341.2093, Δ = −2.7 ppm).ε) 257 (0.97) nm; IR (KBr) νmax 3076, 1807, 1675, 1604, 1252, 1131, 962 cm−1; 1H and 13C NMR data, see Tables 1 and 3; (+) ESIMS m/z 313 [M + H]+, 335 [M + Na]+; HRESIMS m/z 335.1629 [M + Na]+ (calcd for C20H24O3Na, 335.1623, Δ = −3.9 ppm).ε) 300 (3.90) nm; IR (KBr) νmax 2920, 2849, 1780, 1671, 1610, 1452, 1384, 1205, 1177, 1134, 1046, 990, 931, 805, 750 cm−1; 1H and 13C NMR data, see Tables 1 and 3; (+) ESIMS m/z 315 [M + H]+, 337 [M + Na]+, 629 [2M + H]+; HRESIMS m/z 337.1789 [M + Na]+ (calcd for C20H26O3Na, 337.1780, Δ = −4.6 ppm).ε) 217 (0.91) nm; IR (KBr) νmax 3462, 2960, 1721, 1696, 1645, 1442, 1211, 906 cm−1; 1H and 13C NMR data, see Tables 1 and 3; (+) ESIMS m/z 349 [M + H]+, 371 [M + Na]+; HRESIMS m/z 371.1831 [M + Na]+ (calcd for C20H28O5Na, 371.1834, Δ = −0.5 ppm).ε) 243 (1.46), 230 (0.97) nm; IR (KBr) νmax 3456, 2962, 2856, 1723, 1692, 1630, 862 cm−1; 1H and 13C NMR data, see Tables 1 and 3; (+) ESIMS m/z 351 [M + H]+, 373 [M + Na]+; HREIMS m/z 373.1993 [M + Na]+ (calcd for C20H30O5Na, 373.1991, Δ = −1.8 ppm).ε) 296 (1.68), 244 (1.56) nm; IR (KBr) νmax 2927, 2872, 1696, 1459, 1374, 1271, 1138, 952, 866 cm−1; 1H and 13C NMR data, see Tables 2 and 3; (+) ESIMS m/z 333 [M + H]+, 355 [M + Na]+, 687 [2M + Na]+; HRESIMS m/z 333.2071 [M + H]+ (calcd for C20H29O4, 333.2060, Δ = +3.1 ppm).ε) 242 (1.02) nm; IR (KBr) νmax 3489, 1714, 1699, 1696, 1621, 1251, 1142, 836 cm−1; 1H and 13C NMR data, see Tables 2 and 3; (+) ESIMS m/z 351 [M + H]+, 373 [M + Na]+, 701 [2M + H]+; HRESIMS m/z 373.1997 [M + Na]+ (calcd for C20H30O5Na, 373.1991, Δ = −3.3 ppm).ε) 254 (1.83) nm; IR (KBr) νmax 3406, 2922, 2866, 2631, 1728, 1684, 1602, 1566, 1471, 1360, 1281, 1211, 1180, 1064, 906, 852, 722 cm−1; 1H and 13C NMR data, see Tables 2 and 3; (+) ESIMS m/z 317 [M + H]+, 339 [M + Na]+; HRESIMS m/z 339.1570 [M + Na]+ (calcd for C19H24O4Na, 339.1572, Δ = −0.9 ppm).ε) 283 (2.23) nm; IR (KBr) νmax 2952, 2876, 2822, 1692, 1596, 1384, 1355, 968 cm−1; 1H and 13C NMR data, see Tables 2 and 3; (+) ESIMS m/z 275 [M + H]+, 297 [M + Na]+; HRESIMS m/z 297.1463 [M + Na]+ (calcd for C17H22O3Na, 297.1467, Δ = −0.8 ppm).ε) 217 (0.83) nm; IR (KBr) νmax 2922, 2857, 1698, 1452, 1442, 1386, 1366, 1024, 786 cm−1; 1H and 13C NMR data, see ref. 29; (+) ESIMS m/z 319 [M + H]+, 341 [M + Na]+; HRESIMS m/z 341.2095 [M + Na]+ (calcd for C20H30O3Na, 341.2093, Δ = −2.5 ppm).ε) 290 (1.72), 242 (1.54) nm; IR (KBr) νmax 2927, 2872, 1696, 1459, 1374, 1271, 1138, 952, 866 cm−1; 1H and 13C NMR data, see Tables 2 and 3; (+) ESIMS m/z 317 [M + H]+, 339 [M + Na]+, 655 [2M + Na]+; HRESIMS m/z 317.2110 [M + H]+ (calcd for C20H29O3, 317.2111, Δ = −0.5 ppm).PTP1B inhibitory activity assay
Recombinant PTP1B catalytic domain was expressed and purified according to a previous report.9,11 The enzymatic activities of the PTP1B catalytic domain were determined at 30 °C by monitoring the hydrolysis of para-nitrophenyl phosphate (p-NPP). The dephosphorylation p-NPP generates product p-NP, which is monitored at an absorbance of 405 nm by the VersaMax microplate reader (Molecular Devices, USA). In a typical 100 μL assay mixture containing 50 mM 3-[N-morpholino]-propanesulfonic acid (MOPS), pH 6.5, 2 mM p-NPP, and 30 nM recombinant PTP1B, activities were continuously monitored and the initial rate of the hydrolysis was determined using the early linear region of the enzymatic reaction kinetic curve. The IC50 was calculated with Prism 4 software (GraphPad Software, San Diego, CA) from the non-linear curve fitting of the percentage of inhibition (% inhibition) versus the inhibitor concentration [I] by using the following equation: % inhibition = 100/[1 + (IC50/[I])k], where k is the Hill coefficient. Oleanolic acid (purity ≥ 98%) was used as the positive control.PTP1B kinetic analysis
The enzyme kinetic analytic assay39 was carried out in a 100 μL system containing 50 mM MOPS at pH 6.5, 30 nM PTP1B, p-NPP in 2-fold dilution from 80 mM, and different concentrations of the inhibitor. In the presence of the competitive inhibitor, the Michaelis–Menten equation is described as 1/v = (Km/[Vmax[S]])(1 + [I]/Ki)+1/Vmax, where Km is the Michaelis constant, v is the initial rate, Vmax is the maximum rate, and [S] is the substrate concentration. The Ki value was obtained by the linear replot of apparent Km/Vmax (slope) from the primary reciprocal plot versus the inhibitor concentration [I] according to the equation Km/Vmax = 1 + [I]/Ki.Acknowledgements
This work was supported by NSFC grants (No. 21472021, 81273401, 81202420), and the National Basic Research Program of China (973 Program, Grant No. 2013CB530700). The authors thank Dr Courtney Starks (Sequoia Sciences, Inc., USA) for her valuable suggestions. The authors are grateful to Mr Bao-Tong Xiao [Peach Valley (Tao-Hua-Chong), Yingshan County, Hubei Province of China] for the plant collection.Notes and references
- (a) S. H. M. Butchart, M. Walpole, B. Collen, A. van Strien, J. P. W. Scharlemann, R. E. A. Almond, J. E. M. Baillie, B. Bomhard, C. Brown, J. Bruno, K. E. Carpenter, G. M. Carr, J. Chanson, A. M. Chenery, J. Csirke, N. C. Davidson, F. Dentener, M. Foster, A. Galli, J. N. Galloway, P. Genovesi, R. D. Gregory, M. Hockings, V. Kapos, J. F. Lamarque, F. Leverington, J. Loh, M. A. McGeoch, L. McRae, A. Minasyan, M. H. Morcillo, T. E. E. Oldfield, D. Pauly, S. Quader, C. Revenga, J. R. Sauer, B. Skolnik, D. Spear, D. Stanwell-Smith, S. N. Stuart, A. Symes, M. Tierney, T. D. Tyrrell, J. C. Vié and R. Watson, Science, 2010, 328, 1164–1168 CrossRef CAS PubMed; (b) N. C. A. Pitman and P. M. Jørgensen, Science, 2002, 298, 989 CrossRef CAS PubMed.
- J. López-Pujol, F.-M. Zhang and S. Ge, Biodiversity and Conservation, 2006, 15, 3983–4026 CrossRef.
- (a) L.-K. Fu and J.-M. Jin, China Plant Red Data Book. Rare and Endangered Plants I, Science Press, Beijing and New York, 1992 Search PubMed; (b) The State Forestry Administration and the Ministry of Agriculture, List of Wild Plants of National Priority Protection I, 1999, http://www.gov.cn/gongbao/content/2000/content_60072.ht.
- D. J. Newmen and G. M. Cragg, J. Nat. Prod., 2012, 75, 311–335 CrossRef PubMed.
- (a) M. A. Ibrahim, M. Na, J. Oh, R. F. Schinazi, T. R. McBrayer, T. Whitaker, R. J. Doerksen, D. J. Newman, L. G. Zachos and M. T. Hamann, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16832–16837 CrossRef CAS PubMed; (b) F. Zhu, C. Qin, L. Tao, X. Liu, Z. Shi, X. Ma, J. Jia, Y. Tan, C. Cui, J. Lin, C. Tan, Y. Jiang and Y. Chen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 12943–12948 CrossRef CAS PubMed.
- W.-J. Zheng and L.-G. Fu, in Flora of China, Science Press, Beijing, 1978, vol. 7, pp. 32−211 Search PubMed.
- Jiangsu New Medical College, Traditional Chinese Medicine Dictionary, Shanghai Science and Technology Press, Shanghai, 1996, vol. 1, pp. 1252–1259 Search PubMed.
- B. Li, Y.-H. Shen, Y.-R. He and W.-D. Zhang, Chem. Biodiversity, 2013, 10, 2133–2160 Search PubMed.
- C.-L. Hu, J. Xiong, J.-Y. Li, L.-X. Gao, W.-X. Wang, K.-J. Cheng, G.-X. Yang, J. Li and J.-F. Hu, Eur. J. Org. Chem., 2016, 1832–1835 CrossRef CAS.
- (a) M. Elchebly, P. Payette, E. Michaliszyn, W. Cromlish, S. Collins, A. L. Loy, D. Normandin, A. Cheng, J. H. Hagen, C. C. Chan, C. Ramachandran, M. J. Gresser, M. L. Tremblay and B. P. Kennedy, Science, 1999, 283, 1544–1548 CrossRef CAS PubMed; (b) T. O. Johnson, J. Ermolieff and M. R. Jirousek, Nature, 2002, 1, 696–709 CAS; (c) A. Alonso, J. Sasin, N. Bottini, I. Friedberg, I. Friedberg, A. Osterman, A. Godzik, T. Hunter, J. Dixon and T. Mustelin, Cell, 2004, 117, 699–711 CrossRef CAS PubMed.
- J. Xiong, Z.-L. Hong, L.-X. Gao, J. Shen, S.-T. Liu, G.-X. Yang, J. Li, H.-Q. Zeng and J.-F. Hu, J. Org. Chem., 2015, 80, 11080–11085 CrossRef CAS PubMed.
- (a) J. S. Calderón, L. Quijano, F. Gómez-Garibay, M. Morán and T. Rios, Phytochemistry, 1987, 26, 2639–2641 CrossRef; (b) J.-M. Fang, K.-C. Hsu and Y.-S. Cheng, Phytochemistry, 1989, 28, 1173–1175 CrossRef CAS.
- L. J. Gough and J. S. Mills, Phytochemistry, 1974, 13, 1612–1613 CrossRef CAS.
- X. Yang, Y. Ding, Z.-H. Sun and D.-M. Zhang, Acta Pharmaceutica Sinica, 2005, 40, 435–437 CAS.
- X. Yang, H. Zhang, Y.-C. Zhang, Y. Ma and J. Wang, Fitoterapia, 2008, 79, 179–181 CrossRef CAS PubMed.
- S. F. H. Zaidi, S. Awale, S. K. Kalauni, Y. Tezuka, H. Esumi and S. Kadota, Planta Med., 2006, 72, 1231–1234 CrossRef CAS PubMed.
- X.-W. Yang, L. Feng, S. M. Li, X.-H. Liu, Y.-L. Li, L. Wu, Y.-H. Shen, J.-M. Tian, X. Zhang, X.-R. Liu, N. Wang, Y.-H. Liu and W.-D. Zhang, Bioorg. Med. Chem., 2010, 18, 744–754 CrossRef CAS PubMed.
- X.-W. Yang, S.-M. Li, L. Feng, Y.-H. Shen, J.-M. Tian, X.-H. Liu, H.-W. Zeng, C. Zhang and W.-D. Zhang, Tetrahedron, 2008, 64, 4354–4362 CrossRef CAS.
- R. Tanaka, H. Ohtsu and S. Matsunaga, Phytochemistry, 1997, 46, 1051–1057 CrossRef CAS.
- B. Gigante, M. J. Marcelo-Curto, A. M. Lobo and S. Prabhakar, J. Nat. Prod., 1989, 2, 85–94 CrossRef.
- H. T. A. Cheung, T. Miyase, M. P. Lenguyen and M. A. Smal, Tetrahedron, 1993, 49, 7903–7915 CrossRef CAS.
- H. A. Mohamed, C.-L. Hiseh, C. Hsu, C.-C. Kuo, H.-S. Chang, C.-K. Lee, T.-H. Lee, J.-B. Wu, C.-I. Chang and Y.-H. Kuo, Helv. Chim. Acta, 2014, 97, 1146–1151 CrossRef.
- J. P. Teresa, A. F. Barrero, L. Muriel, A. S. Feliciano and M. Grande, Phytochemistry, 1980, 19, 1153–1156 CrossRef.
- Y.-H. Kuo, M.-H. Yeh and H.-C. Lin, Chem. Pharm. Bull., 2004, 52, 861–863 CrossRef CAS PubMed.
- T. A. van Beek, F. W. Claassen, J. Dorado, M. Godejohann, R. Sierra-Alvarez and J. B. P. A. Wijnberg, J. Nat. Prod., 2007, 70, 154–159 CrossRef CAS PubMed.
- Y. Kinouchi, H. Ohtsu, H. Tokuda, H. Nishino, S. Matsunaga and R. Tanaka, J. Nat. Prod., 2000, 63, 817–820 CrossRef CAS.
- T. Ohmoto, K. Kanatani and K. Yamaguchi, Chem. Pharm. Bull., 1987, 35, 229–234 CrossRef CAS.
- M. Stavri, A. Paton and B. W. S. Skelton, J. Nat. Prod., 2009, 72, 1191–1194 CrossRef CAS PubMed.
- S. Janocha, J. Zapp, M. Hutter, M. Kleser, J. Bohlmann and R. Bernhardt, ChemBioChem, 2012, 14, 467–473 CrossRef PubMed.
- S.-B. Wu, Q.-Y. Bao, W.-X. Wang, Y. Zhao, G. Xia, Z. Zhao, H. Zeng and J.-F. Hu, Planta Med., 2010, 76, 1–7 CrossRef.
- W. X. Jiao, J. W. Blunt, A. L. J. Cole and M. H. G. Munro, J. Nat. Prod., 2004, 67, 1434–1437 CrossRef CAS PubMed.
- (a) C. Li, D. Lee, T. N. Graf, S. S. Phifer, Y. Nakanishi, S. Riswan, F. M. Setyowati, A. M. Saribi, D. D. Soejarto, N. R. Farnsworth, J. O. Falkinham III, D. J. Kroll, A. D. Kinghorn, M. C. Wani and N. H. Oberlies, J. Nat. Prod., 2009, 72, 1949–1953 CrossRef CAS PubMed; (b) J.-M. Fang, C.-K. Lee and Y.-S. Cheng, J. Nat. Prod., 1993, 33, 1169–1172 CAS.
- J. G. Luis and T. A. Grillo, Tetrahedron, 1993, 49, 6277–6284 CrossRef CAS.
- H. Ohtsu, R. Tanaka, Y. In, S. Matsunaga, H. Tokuda and H. Nishino, Planta Med., 2001, 67, 55–60 CrossRef CAS PubMed.
- R. N. Moore and R. V. Lawrence, U.S. Patent, 2,899,463, 1959.
- (a) D. Bradley, ChemViews Magazine, 2015 DOI:10.1002/chemv.201500097; (b) L.-J. Wang, J. Xiong, S.-T. Liu, L.-L. Pan, G.-X. Yang and J.-F. Hu, J. Nat. Prod., 2015, 78, 1635–1646 CrossRef CAS PubMed.
- (a) M. Na, W. K. Oh, Y. H. Kim, X. F. Cai, S. Kim, B. Y. Kim and J. S. Ahn, Bioorg. Med. Chem. Lett., 2006, 16, 3061–3064 CrossRef CAS PubMed; (b) S. Kim, M. Na, H. Oh, J. Jang, C. B. Sohn, B. Y. Kim, W. K. Oh and J. S. Ahn, J. Enzyme Inhib. Med. Chem., 2006, 21, 379–383 CrossRef CAS PubMed; (c) H. J. Jung, H. A. Jung, S. S. Kang, J.-H. Lee, Y. S. Cho, K. H. Moon and J. S. Choi, Arch. Pharmacal Res., 2012, 35, 1771–1777 CrossRef CAS PubMed; (d) H. A. Jung, Y. S. Cho, S. H. Oh, S. Lee, B.-S. Min and J. S. Choi, Arch. Pharmacal Res., 2013, 36, 957–965 CrossRef CAS PubMed.
- (a) R.-X. Tan and W.-X. Zou, Nat. Prod. Rep., 2001, 18, 448–459 RSC; (b) S. Kusari, S. P. Pandey and M. Spiteller, Phytochemistry, 2013, 91, 81–87 CrossRef CAS PubMed.
- W. Zhang, D. Hong, Y. Zhou, Y. Zhang, Q. Shen, J.-Y. Li, L.-H. Hu and J. Li, Biochim. Biophys. Acta, 2006, 1760, 1505–1512 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The 1D-/2D NMR, ECD and HRESIMS spectra of compounds 1–11, and spectroscopic data of the known compounds 12–39. See DOI: 10.1039/c6ra08986k |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |