
Preparation of platinum nanoparticles immobilized on ordered mesoporous Co3O4–CeO2 composites and their enhanced catalytic activity†
Qianli Wanga,
Yiwei Zhang*a,
Yuming Zhou*a,
Zewu Zhangb,
Yuanmei Xua,
Chao Zhanga,
Hongxing Zhanga and
Xiaoli Shenga
aSchool of Chemistry and Chemical Engineering, Southeast University, Nanjing 211189, China. E-mail: zhangchem@seu.edu.cn; ymzhou@seu.edu.cn; Fax: +86 25 52090617; Tel: +86 25 52090617
bSchool of Materials Engineering, Nanjing Institute of Technology, Nanjing 211167, China
First published on 29th June 2016
Abstract
Using mesoporous silica KIT-6 as a template, ordered mesoporous Co3O4–CeO2 composites with different contents of cobalt were prepared via the hard template method. Uniform Pt nanoparticles stabilized by polyamidoamine (PAMAM) dendrimers were immobilized on different Co3O4–CeO2 composites, resulting in Pt-based supported catalysts. The prepared materials were characterized by several techniques such as transmission electron microscopy (TEM), nitrogen physical adsorption, scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), X-ray diffraction (XRD) and energy dispersion X-ray analysis (EDX). The results showed that the Co3O4–CeO2 composites have a regular pore structure and high crystallinity. Moreover, the specific surface area of Co3O4–CeO2 reached 135.04 m2 g−1 for 10 mol% of cobalt (Co/(Co + Ce)). The introduction of cobalt could improve the structure of the supports and enhance the catalytic efficiency for the prepared Pt-based supported catalysts. The catalytic performances were evaluated by the reduction of 4-nitrophenol as monitored by UV-Vis spectra. In a comparison of the catalysts with different cobalt contents, it was found that Pt/meso-CeO2Co10 possessed the highest catalytic performance as well as good reusability.
1. Introduction
Platinum nanocatalysts with a small size have been widely applied to catalysis such as in the selective oxidation of alcohols, propane dehydrogenation and the reduction of nitro compounds.1–3 However, naked platinum nanoparticles (Pt NPs) facilely aggregate to form big particles owing to their high surface energy, which decreases their catalytic activity.4 To relieve the aggregation, different templating agents have been employed to stabilize the metallic particles such as surfactants, ligands and dendrimers.5–7 In particular, dendrimers possessing a three-dimensional, highly branched and well-defined structure have been used as a promising templating agent to prepare homogeneously distributed metal NPs.8,9 Unfortunately, the obtained homogeneous nanocatalysts based on the dendrimers encountered the problem of a difficult recovery in reactions. However, the introduction of solid carriers for the catalytic system can facilely overcome this separation problem. Moreover, it can also stabilize and sustain small metal particles depending on the strong interaction between the metal particles and carriers.10 For instance, Ivanova et al. investigated this interaction with Pt/γ-Al2O3 and Pd/γ-Al2O3, which revealed that the metal particles were well dispersed on the support based on the interaction between them.11 In general, the morphology of the carriers largely influences the distribution of metal particles and the catalytic activity. For example, Ye and co-workers prepared the three-dimensional urchin-like WO3-supported metal nanoparticles with a narrow size distribution, and they found this unique morphology of the support distinctly improved the metallic dispersion compared to a traditional WO3 support.12Recently, mesoporous transition metal oxides have attracted considerable interest owing to their unique pore structure, high crystalline quality and large surface area.13,14 Considering the difficult control of hydrolysis of metal precursors and the fragile thermal stability resulting from fabrication with a soft method, mesoporous metal oxides are usually prepared by a hard template method.15–17 For example, Ren et al. successfully prepared different ordered mesoporous metal oxides (e.g., CuO, Fe2O3, Mn2O3, CeO2 and NiO) using mesoporous silica KIT-6 as a hard template, and these exhibited higher catalytic activity for CO oxidation than their bulk counterparts.18 The regular porous structure can enlarge the surface area, provide more active sites and facilitate the adsorption and diffusion of reactant molecules. Based on the beneficial features of mesoporous metal oxides, they are frequently employed to synthesize heterogeneous catalysts.19,20 In particular, as an important rare earth oxide, mesoporous ceria (CeO2) has been widely applied to catalysis and energy applications owing to its unique physicochemical features such as the redox property, high thermal stability and high oxygen storage capacity.21,22 For example, Chong et al. reported Au nanoclusters were deposited on mesoporous ceria nanospheres, exhibiting the efficient catalytic reduction of nitrobenzene compared to catalyst systems with silica or ferric oxide supports.23 Our previous investigation showed Pt NPs supported on ordered mesoporous ceria exhibited better catalytic performance compared to the common ceria support system.24
With the developing of new compound materials in recent years, more attention has been paid to the fabrication of mesoporous ceria oxide doped by other metal oxides, which could improve catalytic activity owing to the formation of heterojunctions.25 For example, Wang et al. reported that mesoporous CeO2–TiO2 composites, as a photocatalyst, possessed higher efficiency in comparison with sole TiO2 or CeO2 for the reduction of CO2 with H2O.26 Tang et al. prepared mesoporous ceria doped by NiO, which displayed a better performance for CO oxidation as the introduction of nickel probably aroused synergetic catalysis.27 Although there are several reports involving mesoporous ceria doped with metal oxides, these composites are often directly used as catalysts in reactions. Actually, as vital functional materials, ceria-based composites still possess special research significance and broad applications. Cobalt oxide (Co3O4) is emerging as an efficient catalyst and as a support for metals for various catalytic processes due to its light harvesting and electron-mediating properties.28 For instance, Co3O4 is a promising catalyst for CO oxidation, VOC oxidation and for the photocatalytic degradation of organic pollution.29,30 Moreover, the investigation from Somorjai et al. revealed that Co3O4 has a stronger synergistic effect with Pt NPs compared with some other oxides such as NiO and MnO2.31 Consequently, the introduction of cobalt to the platinum–ceria catalytic system would promote their synergistic effect, improve the stabilization of metal particles and the structure of the catalyst, and enhance the catalytic activity.
Conventionally, 4-nitrophenol (4-NP) is one of common organic pollutants present in industrial and agricultural wastewater; however, 4-aminophenol (4-AP) is a vital intermediate for the manufacture of a diverse range of analgesic and antipyretic drugs, and anticorrosion lubricants.32 Thus, many researchers have devoted efforts to synthesizing efficient catalysts to promote this conversion. Traditionally, 4-NP is facilely reduced by NaBH4 in the presence of supported noble metal catalysts such as Au/CeO2, Au/Al2O3, Pt/CeO2, Au/grapheme, Ag/TiO2, or Pt/Fe3O4.33–37 However, in the present investigations involving the reduction of 4-NP, little attention has been paid to mesoporous cobalt–ceria composites as supports. However, the unique features of the mesoporous cobalt–ceria material have inspired us to prepare an ordered mesoporous cobalt-ceria composites-supported Pt catalyst for enhancing the reduction of 4-NP.
Herein, we employed mesoporous silica as a template to prepare ordered mesoporous Co3O4–CeO2 composites. Dendrimers were used to synthesize dispersed Pt NPs and then were loaded on the Co3O4–CeO2 composites, which catalyzed the reduction of 4-NP. The synthesis process is depicted in Fig. 1. The utilization of dendrimers improved the dispersion of Pt NPs due to their unique hyperstructure. The Co3O4–CeO2 composites support would promote the electron transition and enhance the catalytic activity, as well as stabilizing the Pt NPs on the carrier by their strong interaction.
 | ||
| Fig. 1 Schematic of the synthesis of platinum immobilized on mesoporous Co3O4–CeO2 composites. | ||
2. Experimental
2.1 Materials
Cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O, ≥99%), cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, ≥98.5%), potassium tetrachloroplatinate (K2PtCl4, ≥96%), ethanol, n-butanol, hydrochloric acid (36–38%), tetraethyl orthosilicate (TEOS, 98%), sodium hydroxide, and sodium borohydride were all purchased from Sinopharm Chemical Reagent Co. Ltd. G4-OH PAMAM dendrimers were purchased from Chen Yuan Molecular New Materials Co., Ltd (Weihai, China), and prior to use, were diluted to 0.34 mM with deionized water. P123 (MW = ∼5800) and 4-nitrophenol (≥99%) were obtained from Sigma Aldrich Corporation.2.2 Preparation of the catalyst
2.3 Characterization of the catalysts
Transmission electron microscopy (TEM) micrographs were obtained from a FEI Tecnai G20 microscope operated at 100 kV. X-ray diffraction (XRD) data were collected on a Bruck D8 instrument using Cu Kα radiation (λ = 1.5406 Å). Field emission scanning electron microscopy (SEM) was performed on a scanning electron microscope (Zeiss, Ultra Plus) unit operating at 20 kV. N2 physical adsorption was performed at 77 K using an ASAP 2020 device (Micrometrics USA). X-ray photoelectron spectroscopy (XPS) was acquired using an Axis Ultra DLD spectrometer with an Al Kα (mono) anode at energy of 150 W. UV-Vis spectra were obtained on a Shimadzu UV 3600 spectrometer. The Pt content of the samples was determined by means of an inductivity coupled plasma mass spectrometry (ICP-MS, Thermo Elemental X7 series).2.4 Catalytic reaction
The catalytic performances of the prepared catalysts were evaluated using the reduction of 4-nitrophenol with NaBH4. In the experiment, 4-nitrophenol (30 μL, 0.01 M) was added to water (1.8 mL), and then this was mixed with NaBH4 ice-solution (1 mL, 0.25 M) in a quartz cell. Then, a dispersed aqueous solution of the catalyst (0.3 mL, 0.5 g L−1) was added to the mixture. The reaction process was monitored with a UV-Vis spectrophotometer at a regular time to measure the decrease in the reaction mixture at 400 nm. For the recycling experiments, the catalysts were collected by centrifugation, washed with water, dried and reused in the next cycles.3. Results and discussions
3.1 Characterization of the catalysts
The pore structure of the as-synthesized silica template (KIT-6) was analyzed via the nitrogen physical adsorption method. In Fig. 2a, it can be observed that the N2 adsorption–desorption isotherm of KIT-6 exhibited a type-IV isotherm with an obvious H1 hysteresis loop, suggesting a typical mesoporous structure on the basis of IUPAC.40 In particular, the sharp inflection at a relative pressure in the range of 0.7–0.8, corresponding to the capillary condensation of N2, indicated the uniform pore structure. The BET surface area of KIT-6 was 717.07 m2 g−1. The pore size distribution of KIT-6 was calculated from the desorption branch using the BJH method, exhibiting quite narrow distribution with a major pore size of around 7.5 nm. In Fig. 2b, the represented TEM image of KIT-6 showed a regular three-dimensional mesoporous structure, which was also confirmed by small-angle XRD of KIT-6 (see the following discussion). The measured pore size was about 7 nm, which was in agreement with the nitrogen physisorption results.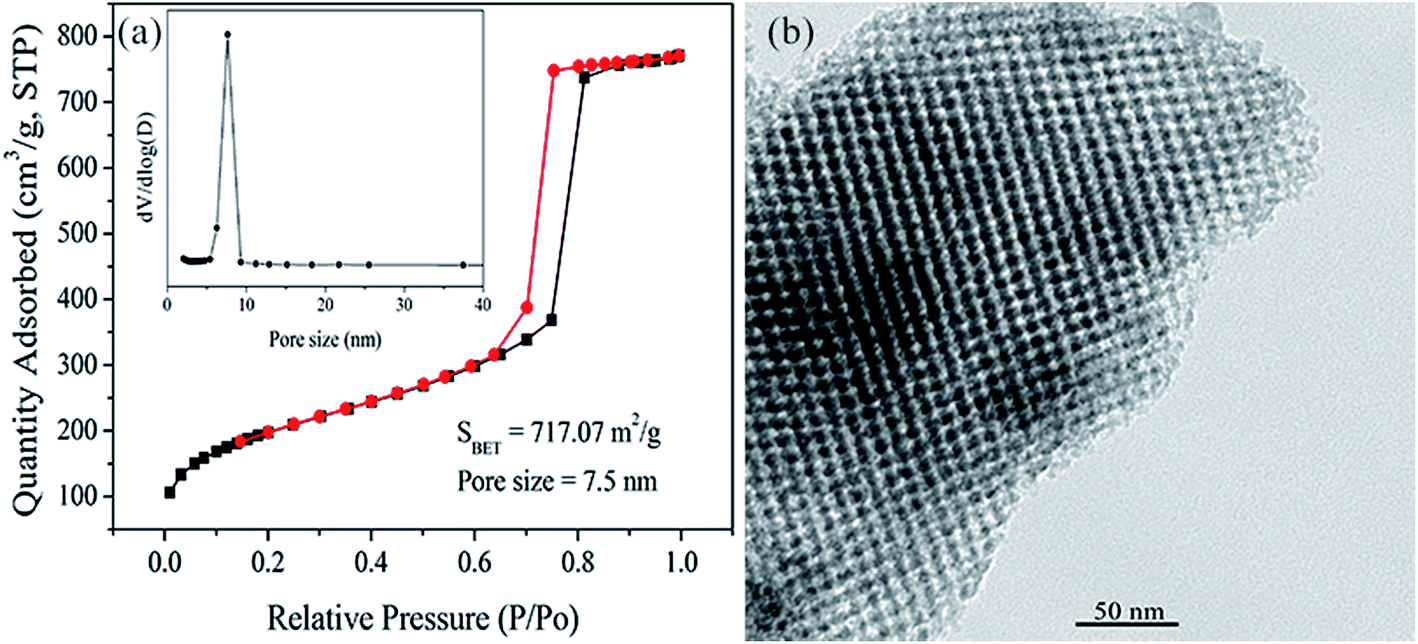 | ||
| Fig. 2 (a) N2 adsorption–desorption isotherms (inset is the corresponding BJH pore size distribution curve) and (b) TEM image of KIT-6. | ||
The regular structure and nanocrystalline structure of the as-prepared materials were determined by small-angle and wide-angle XRD patterns. Fig. 3A presents the small-angle XRD patterns of the parent template KIT-6 and of the ceria-based mesoporous oxides (inset in Fig. 3A). KIT-6 showed three obvious peaks, indexed as (200), (220) and (332) reflections, which correspond to the cubic Ia3d space group.41,42 Based on the regular structure of KIT-6, the introduction of cerium and cobalt salts and the subsequent removal of silica were expected to inversely replicate the ordered structure of KIT-6. According to the inset in Fig. 3A, it was found that ceria-based mesoporous materials all showed a weak diffraction peak at around 2θ = 1° compared with KIT-6, indicating the prepared mesoporous metal oxides mostly inherited the ordered mesoporous structure of their template. Moreover, we also noted that the diffraction peak of the metal oxides was slightly shifted to a higher angle, implying a small shrinkage of the mesoporous structure after replication.43 As presented in Fig. 3B, all the samples exhibited good crystallization and the main phase of cubic fluorite ceria, as well as distinct diffraction peaks at 2θ = 28.6°, 33.2°, 47.4°, 56.4°, 59.1°, 69.4° and 76.8°, which corresponded to the (111), (200), (220), (311), (222), (400) and (331) planes of the ceria phase (JCPDS PDF# 43-1002). When the cobalt species were introduced into CeO2, as presented in Fig. 3B(b), the characteristic diffraction peaks of CeO2 became weak, suggesting a decrease in ceria sizes. Moreover, partial new diffraction peaks emerged at 2θ = 31.3° and 36.9°, which were indexed as the (220) and (311) diffraction planes of Co3O4 (JCPDS PDF# 74-1657), respectively. As for the absence of other diffraction peaks, this was due to the good dispersion and the lower contents of cobalt, which was also observed in similar reports.44 With adding cobalt contents, as shown in the XRD pattern of meso-CeO2Co30 (Fig. 3B(c)), the diffraction peaks of Co3O4 became strong and were clearly displayed at 2θ = 19.1°, 31.3°, 36.9°, 45.0°, 59.3° and 65.4°, which were assigned to the (111), (220), (311), (400), (511), and (440) planes of Co3O4 (JCPDS PDF# 74-1657), respectively, confirming the existence of crystalline Co3O4. Actually, the color of the sample also changed from yellow to brown after the addition of a low amount of cobalt, and the brown color gradually deepened with increasing the cobalt content. In addition, the mean crystalline size of CeO2 was calculated by Scherrer equation based on the CeO2 (111) peak, which is depicted in Table 1. Obviously, the addition of less cobalt would decrease the crystalline size of ceria, and consequently, meso-CeO2Co10 revealed the smallest crystalline size. The small crystallite size also increases the number of reactive edge sites.
 | ||
| Fig. 3 (A) Small-angle XRD patterns of KIT-6 and the mesoporous metal oxide (inset) samples; (B) wide-angle XRD patterns of (a) meso-CeO2, (b) meso-CeO2Co10, and (c) meso-CeO2Co30. | ||
To investigate the textural properties of ceria-based mesoporous metal materials, N2 adsorption–desorption analysis was performed. As revealed in Fig. 4A, the pure meso-CeO2 and meso-Co3O4–CeO2 composites all showed a type-IV sorption isotherm, implying the formation of a mesoporous structure in theses samples. After the introduction of cobalt, the adsorption in the range of high pressure gradually decreased, suggesting a decrease in the rate of interstitial pores between the particles. In Fig. 4B, the pure meso-CeO2 showed a narrow pore size at around 3.28 nm and a relatively wide pore size at around 12 nm. In addition, it was noted that the introduction of cobalt resulted in a narrower pore size distribution compared with pure ceria. This result demonstrated that the metal precursors were well filled in the two sets of pores of KIT-6 with the assistance of the cobalt species, which was similar to the report by Tang et al.27 The textural parameters of the samples are presented in Table 1. meso-CeO2Co10 and meso-CeO2Co30 possessed a specific surface area of 135.04 m2 g−1 and 123.19 m2 g−1, respectively, which are both larger than that (111.30 m2 g−1) of meso-CeO2. Moreover, the specific surface area of meso-Co3O4–CeO2 was also larger than for the flower-like Co3O4–CeO2 composites (42.00 m2 g−1),45 macroporous Ce–Zr oxide (72.60 m2 g−1),46 and mesoporous NiO–CeO2 composites (98.00 m2 g−1),27 indicating the applicability of the hard template method. The large surface area for the supports was helpful in improving the dispersion of metal particles.
 | ||
| Fig. 4 (A) N2 adsorption–desorption isotherms and (B) the corresponding pore size distribution curves of the different samples: (a) meso-CeO2, (b) meso-CeO2Co10, and (c) meso-CeO2Co30. | ||
TEM characterization was carried out to ascertain the morphology of the ceria-based materials. As shown in Fig. 5, all the prepared samples possessed a relatively regular mesoporous structure, which was in good accordance with the small-angle XRD analysis. For meso-CeO2Co10 and meso-CeO2Co30, it was observed that their mesoporous structure was more uniform and the pore size was smaller compared with pure meso-CeO2. meso-CeO2Co10 and meso-CeO2Co30 displayed a preferable spherical structure, which was confirmed by the following SEM analysis. For ceria-based support materials, the good crystallinity played an important role in improving the catalytic activity; therefore, a suitable calcination temperature was important. In the process of decomposition of the mixture of ceria salt and cobalt salt, TG analysis showed that a majority of the precursors decomposed at 300 °C, with complete decomposition at 500 °C, as shown in Fig. S2.† As a result, 500 °C was chosen as the final calcination temperature. The selected-area electron diffraction (SAED) patterns, as observed in the inset in Fig. 5, showed multiple bright diffraction rings, implying the high crystallinity of Co3O4–CeO2 composites, which was in agreement with the wide-angle XRD analysis. Moreover, it was noted that the SAED patterns of meso-CeO2Co30 (inset in Fig. 5c) obtained a dim ring corresponding to the (220) plane of Co3O4, implying the presence of Co3O4. To closely study the structure of the obtained samples, meso-CeO2Co30 was chosen as the example to be analyzed by high-resolution TEM (Fig. 5d). Lattice spacing d values of 0.35 nm and 0.28 nm were clearly observed, corresponding to the (111) plane of CeO2 (JCPDS PDF# 43-1002) and the (220) plane of Co3O4 (JCPDS PDF# 74-1657), thus confirming the high crystallinity and the existence of ceria and cobalt. As presented in Fig. S2,† the EDX analysis of meso-CeO2Co10 and meso-CeO2Co30 proved that the prepared composites contained Ce and Co species. The corresponding ratio data are depicted in Table 1.
 | ||
| Fig. 5 Low-magnitude TEM images of (a) meso-CeO2, (b) meso-CeO2Co10 and (c) meso-CeO2Co30 (insets are the corresponding SAED patterns); (d) high-resolution TEM image of meso-CeO2Co30 (inset is the corresponding high-magnitude image). | ||
The prepared mesoporous materials were used as supports to load Pt NPs, which were characterized by TEM. Herein, uniform Pt NPs were prepared previously in solution depending on the stabilization by the PAMAM dendrimers. Fig. S3† reveals uniformly dispersed Pt NPs with a small particle size of 1.81 ± 0.09 nm, indicating the availability of the PAMAM dendrimers. Fig. 6 displays the TEM images of Pt/meso-CeO2, Pt/meso-CeO2Co10 and Pt/meso-CeO2Co30, which reveal that the prepared catalysts basically preserved a good mesoporous structure and a relatively homogeneous distribution of Pt NPs. The size of the Pt NPs was around 3.52 nm for Pt/meso-CeO2Co10, whereas the Pt NPs on meso-CeO2 exhibited sizes of about 4.15 nm. Moreover, we also noted that the mesoporous structure suffered partial destruction compared with the original supports, maybe because the weak acid solution influenced the pore structure of supports in the loading process. In particular, meso-CeO2Co30 showed a relatively bad mesoporous structure after loading Pt NPs, which was because the large cobalt oxide was facilely etched by acid solution. Consequently, a suitable cobalt content is necessary for sustaining the pore structure of the catalysts. In general, a low ordered structure is not conducive to the dispersion of metal nanoparticles, which was confirmed by partial aggregation of the Pt NPs and by the increase in particle size of Pt NPs, with a size of around 5.02 nm. The corresponding EDX analysis in Fig. 6d–f showed the presence of platinum, implying that Pt NPs were successfully immobilized on ceria-based supports. For Pt/meso-CeO2, Pt/meso-CeO2Co10 and Pt/meso-CeO2Co30, the Pt loading tested by ICP-MS was 1.6 wt%, 1.3 wt% and 1.5 wt%, respectively, suggesting little distinction for the different Pt contents.
 | ||
| Fig. 6 (a–c) TEM images of Pt/meso-CeO2, Pt/meso-CeO2Co10 (inset is the corresponding SAED pattern) and Pt/meso-CeO2Co30; (d–f) the corresponding EDX analysis patterns. | ||
Fig. 7 presents the SEM images of Pt/meso-CeO2, Pt/meso-CeO2Co10, and Pt/meso-CeO2Co30. In Fig. 7a, it can be observed that Pt/meso-CeO2 showed small particles and some of them were similar to spheres, which were also associated with the TEM analysis. However, meso-CeO2Co10 almost exhibited spherical particles, as shown in Fig. 7b. The locally enlarged image also clearly showed some rough pore structures on the surface of the microspheres, which also confirmed the existence of mesoporous structures. The EDX mapping analysis ascertained the Ce, Co and Pt elements again, and showed a relative homodistribution of Pt NPs. As for the meso-CeO2Co30, it displayed a perfect spherical structure with a distinct pore structure on the surface of the spheres.
 | ||
| Fig. 7 SEM images of (a) Pt/meso-CeO2, (b) Pt/meso-CeO2Co10 (insets are the locally enlarged image and EDX mapping of Ce, Co and Pt elements) and (c) Pt/meso-CeO2Co30. | ||
After the Pt NPs were loaded on the supports, the crystallinity of the prepared catalysts was analyzed by wide-angle XRD patterns. As shown in Fig. 8, all the samples possessed good crystallinity. Moreover, the characteristic diffraction peaks, corresponding to CeO2 and Co3O4, remained unchanged for the obtained catalysts, implying that the loading process did not influence the crystallinity of the supports. However, a new weak diffraction peak at 2θ = 39.6° was noticed, corresponding to the (111) plane of platinum (JCPDS, PDF# 04-0802) for all the catalysts, indicating the successful immobilization of the Pt NPs. The weak intensity was ascribed to the low loading or the uniform dispersion of the Pt NPs. Compared with meso-CeO2 and meso-CeO2Co10, the diffraction peak of Pt NPs for meso-CeO2Co30 showed a slightly stronger and revealed bigger size of Pt NPs, which was in accordance with the TEM characterization.
 | ||
| Fig. 8 XRD patterns of (a) Pt/meso-CeO2, (b) Pt/meso-CeO2Co10 and (c) Pt/meso-CeO2Co30. | ||
XPS is a useful technique to investigate the surface elemental compositions and metal oxidation states of solid materials. Fig. 9 presents the XPS survey spectra of Pt/meso-CeO2Co10. The spectrum of Ce 3d was decomposed into eight components, with the assignment defined in Fig. 9a, where u and v indicate the spin-orbit coupling 3d3/2 and 3d5/2, respectively. The peaks marked as u′′′, u′′, u′, v′′′, v′′and v′ were associated with Ce4+, whereas the u0/v0 doublet was ascribed to the photoemission from Ce3+ cations.47 In Fig. 9b, the Co 2p1/2 and Co 2p3/2 peaks located at around 795 eV and 780 eV demonstrated the existence of Co3O4.48 Co2+ and Co3+ signals were observed after the Co 2p spectra were further deconvolved. Co 2p peaks at about 779 eV and 794 eV corresponded to Co3+, whereas the peaks at 781 eV and 796 eV represented Co2+. As displayed in Fig. 9c, the main peak at around 529 eV represented lattice oxygen, whereas the peak located at about 531 eV was indexed to defective or adsorptive oxygen.47 Fig. 9d shows the Pt 4f spectrum of Pt/meso-CeO2Co10. It was observed that the Pt 4f peaks showed a doublet at around 70.7 eV and 74.0 eV, corresponding to Pt 4f7/2 and Pt 4f5/2. This indicated that Pt was mainly present in the Pt(0) state.49 The strong synergistic interaction between Pt and the ceria–cobalt composites could significantly enhance the catalytic performance.
 | ||
| Fig. 9 XPS spectra corresponding to the Ce 3d, Co 2p, O 1s, and Pt 4f signals of Pt/meso-CeO2Co10. | ||
3.2 Catalytic reaction
The prepared catalysts were employed to catalyze the reduction reaction of 4-NP, with NaBH4 used as the reducing agent. In fact, the reduction of 4-NP has also been widely employed to evaluate the catalytic activity of noble metal-based catalysts.50,51 In general, 4-NP solution possesses a distinct absorption peak at around 317 nm, whereas a new absorption peak appears at 400 nm upon the addition of NaBH4, which originates from the formation of 4-nitrophenolate ion in alkaline solution.52 In the absence of catalyst, the maximum absorption peak of the solution remained unchanged, indicating the reaction hardly proceeded. However, after the Pt-based catalyst was induced, as presented in Fig. 10a, the absorption peak at 400 nm decreased gradually and a new peak centered at 296 nm, corresponding to 4-AP, was observed concomitantly, implying the nitro group of 4-NP was decayed and reduced. Moreover, the color of the solution turned to colorless by degrees during the reaction. In addition, when pure mesoporous ceria support was employed to catalyze the reduction, it was observed that the reaction was processed scarcely, suggesting the reduction was mainly driven by the Pt NPs. | ||
| Fig. 10 (a) Variation in UV-Vis spectra for the 4-NP reduction in the presence of Pt/meso-CeO2Co10; (b) ln(C0/Ct) against the reaction time (t) for the reduction of 4-NP with different samples. | ||
The kinetics of the catalytic reduction of 4-NP in the presence of different catalysts were studied and the results are depicted in Fig. 10b. Considering the excess NaBH4 in the reaction, the concentration of NaBH4 was considered to be stable throughout the reaction, so the catalytic rate was supposed to be evaluated by a pseudo-first-order rate kinetics.53 Fig. 10b reveals the satisfied linear relationship between ln(C0/Ct) against reaction time in the reduction over different prepared samples, demonstrating the reaction obeyed pseudo-first-order kinetics. The rate constant (k) was calculated according to the slope of the fitted line. The obtained rate constants were 6.03 × 10−3 s−1, 9.63 × 10−3 s−1 and 11.30 × 10−3 s−1 for Pt/meso-CeO2, Pt/meso-CeO2Co30, and Pt/meso-CeO2Co10, respectively, indicating the composites with cobalt species significantly enhanced the catalytic performance. This phenomenon was probably attributed to the reasons that the addition of cobalt species improved the pore structure and the special surface area of the supports and cobalt combined with ceria acted as synergistic catalysts.27 In addition, it was observed that Pt/meso-CeO2Co10 showed the highest catalytic activity, which was because the larger surface area and more regular pore structure of meso-CeO2Co10 improved the dispersion of Pt NPs and maintained their small size. Moreover, according to the analysis of XRD, meso-CeO2Co10 had the smaller crystalline size, which facilitated the catalytic efficiency. With the purpose of further comparing the catalytic performance of the as-synthesized catalyst with other Pt-based materials in the literature, the normalized rate constant knor (=k/m, where m was the mass of the catalyst) was calculated. As shown in Table S1,† in this study, the knor value of Pt/meso-CeO2Co10 (75.33 s−1 g−1) was distinctly higher than for some reported Pt-based catalysts, such as Pt nanoflowers (7.80 s−1 g−1), RGO/Pt/CeO2 (11.28 s−1 g−1), Fe3O4@PDA-Pt (57.50 s−1 g−1), PtNi/RGO (43.40 s−1 g−1) and mSiO2/Pt/CeO2/Fe (15.06 s−1 g−1), indicating its superior performance.
As is well known, the reusability is important for heterogeneous catalysts. By experiment, the reusability of the as-prepared Pt/meso-CeO2Co10 catalyst toward the reduction of 4-NP was investigated. As displayed in Fig. 11, the recycling experiment showed no significant decrease in the conversion of 4-NP after five cycles. The conversion yield of 4-NP still sustained the conversion at around 93.5%.
 | ||
| Fig. 11 Conversion of 4-NP in five cycles with Pt/meso-CeO2Co10. | ||
According to the abovementioned experimental results, a presumed reaction mechanism was determined and is provided in Fig. 12. In general, due to the kinetic barriers, the reduction of 4-NP proceeded successfully due to the medium, which was also confirmed by the abovementioned results. Herein, BH4- and 4-NP molecular was adsorbed on the surface of the Pt-based catalyst prior to reaction, and the electron transfer from BH4- to 4-NP was relayed by the Pt NPs.54 Based on Plieth's study, the redox potential decreased with the decreasing size of metal NPs and the smaller Pt NPs could promote electron transfer.55,56 In this experiment, with the addition of a small amount of cobalt species, mesoporous cobalt–ceria composites with a more regular structure and larger surface area were obtained. As revealed in the TEM and BET results, the large surface area of meso-CeO2Co10 improved the dispersion of Pt NPs on the supports. The mesoporous structure also restricted the aggregation of Pt NPs and maintained the small size of metal particles. More importantly, the introduction of Co3O4 decreased the energy barrier and exerted synergistic catalysis when combined with ceria and platinum. However, when a large amount of cobalt was added, the pore structure was partially destroyed owing to the weak acid environment in the loading process. Consequently, the results showed that meso-CeO2Co10 was an ideal support and that Pt/meso-CeO2Co10 exhibited excellent catalytic performance for the reduction of 4-nitrophenol.
 | ||
| Fig. 12 Presumed reaction mechanism for the reduction of 4-nitrophenol over Pt/meso-CeO2Co10 catalyst. | ||
4. Conclusions
In this study, KIT-6 was employed as a template, and meso-CeO2, meso-CeO2Co10 and meso-CeO2Co30 with ordered mesoporous structures and high crystallinity were successfully prepared by the hard template method. In particular, meso-CeO2Co10 possessed a large specific surface area of 135.04 m2 g−1 and a regular pore structure. Uniformly dispersed Pt NPs stabilized by PAMAM dendrimers were loaded on the prepared cobalt–ceria supports. The ordered mesoporous structure prevented the aggregation of Pt NPs, whereas the addition of cobalt species improved the pore structure of the supports. The results revealed Pt/meso-CeO2Co10 exhibited more dispersed Pt NPs with a small size (around 3.45 nm). For the reduction reaction of 4-nitrophenol, Pt/meso-CeO2Co10 exhibited the highest catalytic activity compared with Pt/meso-CeO2 and Pt/meso-CeO2Co30, corresponding to a rate constant of 11.30 × 10−3 s−1. Moreover, Pt/meso-CeO2Co10 also showed good reusability.Acknowledgements
The authors are grateful for the financial supports from the National Natural Science Foundation of China (Grant No. 21376051, 21106017 and 21306023), the Natural Science Foundation of Jiangsu (Grant No. BK20131288), Fund Project for Transformation of Scientific and Technological Achievements of Jiangsu Province of China (Grant No. BA2014100), Fundamental Research Funds for the Central Universities (No. 3207045421) and Instrumental Analysis Fund of Southeast University.References
- Y. W. Zhang, Y. M. Zhou, L. Huang, S. J. Zhou, X. L. Sheng, Q. L. Wang and C. Zhang, Chem. Eng. J., 2015, 270, 352 CrossRef CAS.
- S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17 RSC.
- F. M. Li, X. Q. Gao, Q. Xue, S. N. Li, Y. Chen and J. M. Lee, Nanotechnology, 2015, 26, 065603 CrossRef CAS PubMed.
- X. M. Chen, G. H. Wu, J. M. Chen, X. Chen, Z. X. Xie and X. R. Wang, J. Am. Chem. Soc., 2011, 133, 3693 CrossRef CAS PubMed.
- H. Z. Yang, Y. G. Tang and S. Z. Zou, Electrochem. Commun., 2014, 38, 134 CrossRef CAS.
- S. Alayoglu and B. Eichhorn, J. Am. Chem. Soc., 2008, 130, 17479 CrossRef CAS PubMed.
- P. X. Zhao, N. Li and D. Astruc, Coord. Chem. Rev., 2013, 257, 638 CrossRef CAS.
- M. Pittelkow, K. Moth-Poulsen, U. Boas and J. B. Christensen, Langmuir, 2003, 19, 7682 CrossRef CAS.
- Y. Borodko, C. M. Thompson, W. Y. Huang, H. B. Yildiz, H. Frei and G. A. Somorjai, J. Phys. Chem. C, 2011, 115, 4757 CAS.
- J. A. Farmer and C. T. Campbell, Science, 2010, 329, 933 CrossRef CAS PubMed.
- A. S. Ivanova, E. M. Slavinskaya, R. V. Gulyaev, V. I. Zaikovskii, O. A. Stonkus, I. G. Danilova, L. M. Plyasova, I. A. Polukhina and A. I. Boronin, Appl. Catal., B, 2010, 97, 57 CrossRef CAS.
- G. C. Xi, J. H. Ye, Q. Ma, N. Su, H. Bai and C. Wang, J. Am. Chem. Soc., 2012, 134, 6508 CrossRef CAS PubMed.
- S. M. Sun, W. Z. Wang, S. Z. Zeng, M. Shang and L. Zhang, J. Hazard. Mater., 2010, 178, 427 CrossRef CAS PubMed.
- D. Gu and F. Schuth, Chem. Soc. Rev., 2014, 43, 313 RSC.
- Y. Ren, Z. Ma and P. G. Bruce, Chem. Soc. Rev., 2012, 41, 4909 RSC.
- Y. Ren, Z. Ma and P. G. Bruce, J. Mater. Chem., 2012, 22, 15121 RSC.
- X. H. Sun, R. You, X. D. Hu, J. B. Mo, R. Xiong, H. M. Ji, X. L. Li, S. Cai, C. M. Zheng and M. Meng, RSC Adv., 2015, 5, 35524 RSC.
- Y. Ren, Z. Ma, L. P. Qian, S. Dai, H. Y. He and P. G. Bruce, Catal. Lett., 2009, 131, 146 CrossRef CAS.
- M. S. Jin, J. N. Park, J. K. Shon, J. H. Kim, Z. H. Li, Y. K. Park and J. M. Kim, Catal. Today, 2012, 185, 183 CrossRef CAS.
- Q. Ye, J. S. Zhao, F. F. Huo, D. Wang, S. Y. Cheng, T. F. Kang and H. X. Dai, Microporous Mesoporous Mater., 2013, 172, 20 CrossRef CAS.
- T. Y. Yu, J. Zeng, B. Lim and Y. N. Xia, Adv. Mater., 2010, 22, 5188 CrossRef CAS PubMed.
- P. F. Ji, J. L. Zhang, F. Chen and M. Anpo, J. Phys. Chem. C, 2008, 112, 17809 CAS.
- H. B. Chong, P. Li, J. Xiang, F. Y. Fu, D. D. Zhang, X. R. Ran and M. Z. Zhu, Nanoscale, 2013, 5, 7622–7628 RSC.
- Q. L. Wang, Y. W. Zhang, Y. M. Zhou, Z. W. Zhang, J. J. Xue, Y. M. Xu, C. Zhang, X. L. Sheng and N. S. Kui, RSC Adv., 2016, 6, 730 RSC.
- Y. G. Wang, F. Wang, Y. T. Chen, D. F. Zhang, B. Li, S. F. Kang, X. Li and L. F. Cui, Appl. Catal., B, 2014, 147, 602 CrossRef CAS.
- Y. G. Wang, B. Li, C. L. Zhang, L. F. Cui, S. F. Kang, X. Li and L. H. Zhou, Appl. Catal., B, 2013, 130, 277 CrossRef.
- C. J. Tang, J. C. Li, X. J. Yao, J. F. Sun, Y. Cao, L. Zhang, F. Gao, Y. Deng and L. Dong, Appl. Catal., A, 2015, 494, 77 CrossRef CAS.
- Y. L. Zheng, W. Z. Wang, D. Jiang and L. Zhang, Chem. Eng. J., 2016, 284, 21 CrossRef CAS.
- L. F. Liotta, H. J. Wu, G. Pantaleo and A. M. Venezia, Catal. Sci. Technol., 2013, 3, 3085 CAS.
- J. H. Kou, C. Bennett-Stamper and R. S. Varma, Nanoscale, 2011, 3, 4958 RSC.
- K. An, S. Alayoglu, N. Musselwhite, S. Plamthottam, G. Melaet, A. E. Lindeman and G. A. Somorjai, J. Am. Chem. Soc., 2013, 135, 16689 CrossRef CAS PubMed.
- T. R. Lin, J. Wang, L. Q. Guo and F. F. Fu, J. Phys. Chem. C, 2015, 119, 13658 CAS.
- L. Zhou, X. X. Li, Y. Wang, M. Hong, Y. Y. Liang and J. Zhao, RSC Adv., 2014, 4, 42965 RSC.
- M. M. Mohamed and M. S. Al-Sharif, Appl. Catal., B, 2013, 142, 432 CrossRef.
- J. Li, C. Y. Liu and Y. Liu, J. Mater. Chem., 2012, 22, 8426 RSC.
- Y. M. Xu, Y. W. Zhang, Y. M. Zhou, S. M. Xiang, Q. L. Wang, C. Zhang and X. L. Sheng, RSC Adv., 2015, 5, 58237 RSC.
- X. Huang, X. P. Liao and B. Shi, Green Chem., 2011, 13, 2801–2805 RSC.
- T. W. Kim, F. Kleitz, B. Paul and R. Ryoo, J. Am. Chem. Soc., 2005, 127, 7601 CrossRef CAS PubMed.
- M. R. Knecht, M. G. Weir, V. S. Myers, W. D. Pyrz, H. C. Ye, V. Petkov, D. J. Buttrey, A. I. Frenkel and R. M. Crooks, Chem. Mater., 2008, 20, 5218 CrossRef CAS.
- H. Shankar, G. Rajasudha, A. Karthikeyan, V. Narayanan and A. Stephen, Nanotechnology, 2008, 19, 315711 CrossRef CAS PubMed.
- F. Kleitz, S. H. Choi and R. Ryoo, Chem. Commun., 2003, 2136 RSC.
- P. Visuvamithiran, M. Palanichamy, K. Shanthi and V. Murugesan, Appl. Catal., A, 2013, 462, 31 CrossRef.
- V. Muller, M. Rasp, J. Rathousky, B. Schutz, M. Niederberger and D. Fattakhova-Rohlfing, Small, 2010, 6, 633 CrossRef PubMed.
- L. F. Liotta, G. Di Carlo, G. Pantaleo and G. Deganello, Catal. Commun., 2005, 6, 329–336 CrossRef CAS.
- S. J. Lin, G. J. Su, M. H. Zheng, D. K. Ji, M. K. Jia and Y. X. Liu, Appl. Catal., B, 2012, 123, 440 CrossRef.
- S. X. Cai, D. S. Zhang, L. Zhang, L. Huang, H. R. Li, R. H. Gao, L. Y. Shi and J. P. Zhang, Catal. Sci. Technol., 2014, 4, 93 CAS.
- G. Z. Chen, Q. H. Xu, Y. Yang, C. C. Li, T. Z. Huang, G. X. Sun, S. X. Zhang, D. L. Ma and X. Li, ACS Appl. Mater. Interfaces, 2015, 7, 23538 CAS.
- W. Q. Song, A. S. Poyraz, Y. T. Meng, Z. Ren, S. Y. Chen and S. L. Suib, Chem. Mater., 2014, 26, 4629 CrossRef CAS.
- M. Qamar, M. A. Gondal and Z. H. Yamani, Catal. Commun., 2010, 11, 768 CrossRef CAS.
- J. M. Zhang, G. Z. Chen, D. Guay, M. Chaker and D. L. Ma, Nanoscale, 2014, 6, 2125 RSC.
- X. Wang, D. P. Liu, S. Y. Song and H. J. Zhang, J. Am. Chem. Soc., 2013, 135, 15864 CrossRef CAS PubMed.
- Y. Zhang, Z. M. Cui, L. D. Li, L. Guo and S. H. Yang, Phys. Chem. Chem. Phys., 2015, 17, 14656 RSC.
- Z. W. Zhang, Y. M. Zhou, Y. W. Zhang, S. J. Zhou, S. M. Xiang, X. L. Sheng and P. Jiang, J. Mater. Chem. A, 2015, 3, 4642 CAS.
- J. M. Zheng, Y. L. Dong, W. F. Wang, Y. H. Ma, J. Hu, X. J. Chen and X. G. Chen, Nanoscale, 2013, 5, 4894 RSC.
- W. J. Plieth, J. Phys. Chem., 1982, 86, 3166 CrossRef CAS.
- S. C. Tang, S. Vongehr and X. K. Meng, J. Mater. Chem., 2010, 20, 5436 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra08784a |
| This journal is © The Royal Society of Chemistry 2016 |