
Tunable bioelectrodes with wrinkled-ridged graphene oxide surfaces for electrochemical nitrate sensors†
Abstract
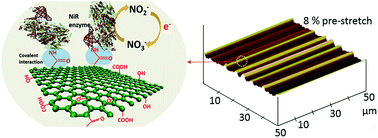
The paper reports on controlled formation of microscale wrinkles and ridges on the surface of a bioelectrode via mechanical stretching to tune and optimize the electrochemical sensing performances of graphene oxide (GO) based nitrate ion sensors. The bioelectrode consists of GO nanosheets drop-coated on a gold (Au) layer with a pre-stretched elastomer substrate. Enzyme nitrate reductase is used for covalent immobilization on the wrinkled-ridged GO surface. Upon relaxation from the pre-stretch, wrinkles or ridges are formed in the GO layer. As the pre-stretch increases, the sinusoidal wrinkles transform to localized ridges on the surface of bioelectrodes. Such morphological transitions, realized by simple mechanical stretching and relaxing, allow optimizing of the electrochemical current and sensing characteristics of the nitrate sensor. The sensing performances of the bioelectrodes at different pre-stretches are investigated. In addition to an increased electroactive surface area, the predominant localized ridges with small sinusoidal wrinkles formed on the GO surface provide a favorable spatial feature, enabling efficient radial diffusion of nitrate ions from surrounding analyte solutions onto the surface of the textured bioelectrode. At the pre-stretch of 8%, the nitrate sensor using the wrinkled-ridged bioelectrode exhibits a considerably high sensitivity of 0.224 μA L mol−1 cm−2 in response to nitrate ions, which is five times higher than that provided by the planar counterpart. Also, the textured bioelectrode shows high selectivity even in the presence of other inferring ions. The present nitrate sensor has potential applications in nitrate detection in sustainable agriculture, environmental monitoring, food analysis, and pharmaceutical industries.
 Please wait while we load your content...
Please wait while we load your content...

