DOI:
10.1039/C6RA08625J
(Paper)
RSC Adv., 2016,
6, 47145-47150
Super/hyperhalogen aromatic heterocyclic compounds†
Received
4th April 2016
, Accepted 1st May 2016
First published on 3rd May 2016
Abstract
Aromatic heterocyclic molecules play an important role in synthetic organic chemistry due to their wide range of reactivities. Because of their aromaticity, they have a lower tendency to accept electrons, and have accordingly been found to have very low and even negative electron affinity values. Superhalogens on the other hand mimic the chemistry of halogens by having electron affinity values higher than that of chlorine, which has the highest electron affinity (at 3.6 eV) of the elements in the periodic table. It is therefore challenging to make superhalogens out of aromatic heterocyclic molecules. In the current work, however, we showed that this is indeed possible by carrying out first-principles calculations. By employing a systematic approach, we were able to achieve superhalogenic aromatic heterocycles having an electron affinity value of 6.00 eV.
Introduction
Superhalogens (SHs)1–8 constitute a category of molecules having electron affinities even higher than those of halogens including chlorine, which has the highest electron affinity of the elements in the periodic table. Due to their highly positive electron affinity, they can be used as powerful oxidizing agents in the synthesis of new compounds. The first superhalogens that were made usually designed by having a metal surrounded by ligands the metal having one excess electron in its valence shell. Since, an appreciable amount of study has been done to model and synthesize advanced superhalogens with a variety of shapes and properties,9–13 by using transition metals at the center14,15 and ligands such as O, O2, NO3, BH4, CN, BO2 and BF4.16–20 A recent study revealed that most of the superhalogens have inorganic rather than organic character. In the current work we have tried computationally to make a set of organic superhalogen molecules from commonly used heterocyclic compounds by following the strategy described by Giri et al.21 In a recent publication, Jena et al.22 showed how one can produce organic superhalogen molecules by functionalizing the benzene molecule. In our present study, we have chosen aromatic heterocyclic compounds because they have been shown, using modern synthetic organic chemistry, to have a wide variety of properties. Generally, they can be divided into two forms: aliphatic and aromatic. The aromatic heterocycles play a major role in drug design for medicinal chemistry. Most natural products have aromatic heterocycles in their structure. Aromatic heterocyclic compounds can be found in biology, pharmacology, optics, and even in the material sciences.23–26 In the current work, we selected eleven commonly used aromatic heterocyclic compounds, and attempted to introduce into them a special property by modifying their structures to a small extent.
We designed our superhalogens by using heterocyclic compounds that are aromatic since they obey Huckel's rule of π-aromaticity.27,28 There are two key issues that we are going to address in this manuscript: firstly, whether aromatic heterocyclic compounds can be transformed into superhalogens; and secondly, what kind of ligand is important to use to make superhalogens. Replacing one C atom with a B atom in the aromatic ring makes the molecule non-aromatic. To regain the aromaticity, such a molecule needs one extra electron to fulfill Huckel's rule of aromaticity. This extra electron makes the molecule aromatic and at the same time switches negative electron affinity to being positive. The strategy we used to make aromatic heterocyclic superhalogens is depicted in Scheme 1. To achieve our goal, we used density functional theory (DFT) to calculate electron affinity (EA), and vertical detachment energy (VDE). Nucleus-independent chemical shift (NICS) calculations were also performed to ensure the aromaticity of the newly developed heterocycles.
 |
| | Scheme 1 Flow chart to prepare superhalogen heterocycles from aromatic heterocycles. | |
Computational details
We calculated electron affinities from the energy of the optimized geometries of neutral and anionic aromatic heterocyclic molecules by using the density functional theory method with the Becke, 3-parameter, Lee–Yang–Parr (B3LYP)29,30 hybrid functional for exchange–correlation and the 6-31+G(d,p) basis set. Frequency calculations were performed using the same level of theory and basis set to ensure the real minima (i.e., Nimag = 0). The geometries were fully optimized without imposing any symmetry constraint. The EA was calculated as the energy difference between the ground state of the anion and that of the corresponding neutral species. Vertical detachment energy (VDE) was determined by calculating the difference between the energy of the ground state anion and that of the corresponding neutral species in the anion geometry. Since global hybrid functionals such as the B3LYP level of calculation are known to overestimate the energy gap, especially when calculating electronic properties such as the ionization potentials, electron affinities, etc. of polyaromatic hydrocarbons,31,32 we re-optimized all of the structures and calculated EA and VDE in the MP2 (ref. 33) level of theory using the same basis set. A benchmark calculation was also performed by taking two molecules from our eleven studied heterocycles in three different newly developed levels of theory, which were based on range-separated functionals such as LC-BLYP,34 LC-wPBE35 and wB97XD.36 The nuclear-independent chemical shifts (NICS)37–39 method was used to evaluate the aromaticity of the compounds. We computed NICS(0) and NICS(1) to make sure that the molecules are aromatic. All of the optimizations and NICS calculations of the heterocyclic molecules were performed using the Gaussian 09 program.40
Results and discussion
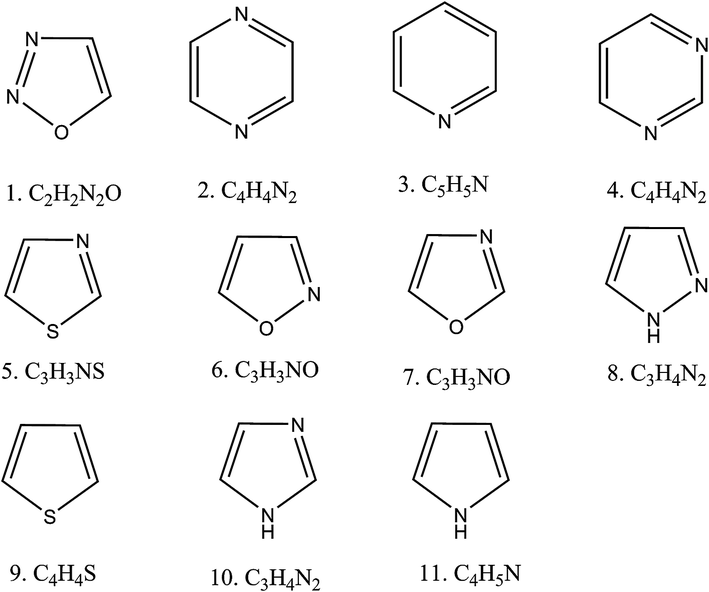
In our study, we selected eleven different aromatic heterocyclic compounds that are commonly used in synthetic organic chemistry. Fig. 1 shows their schematic diagrams. The optimized geometries of their neutral and anionic forms are given in (ESI†). From the optimized energies of the anions and neutral species, we calculated the electron affinities. We found all of the molecules to have negative electron affinities, indicating that it would be unfavorable for them to accept any extra electron. The corresponding EA values are given in Table 1.
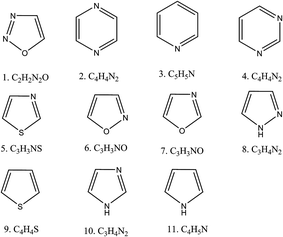 |
| | Fig. 1 Schematic diagram of the aromatic heterocyclic molecules used in this study. | |
Table 1 Electron affinity (EA, eV) and NICS(0,1) values for the aromatic heterocyclic molecules computed at different levels of theory
| Sl. no. |
Aromatic heterocycle formula |
EA (eV) (B3LYP) |
EA (eV) (MP2) |
NICS(0) [ppm] (B3LYP) |
NICS(1) [ppm] (B3LYP) |
| 1 |
C2H2N2O |
−0.333 |
−1.277 |
−13.375 |
−11.458 |
| 2 |
C4H4N2 |
−0.070 |
−0.678 |
−5.341 |
−10.112 |
| 3 |
C5H5N |
−0.749 |
−1.358 |
−6.836 |
−10.012 |
| 4 |
C4H4N2 |
−0.314 |
−1.003 |
−5.545 |
−9.851 |
| 5 |
C3H3NS |
−0.669 |
−1.399 |
−12.714 |
−10.936 |
| 6 |
C3H3NO |
−0.944 |
−1.722 |
−12.133 |
−10.297 |
| 7 |
C3H3NO |
−1.139 |
−1.848 |
−11.480 |
−9.659 |
| 8 |
C3H4N2 |
−1.056 |
−1.353 |
−13.799 |
−11.284 |
| 9 |
C4H4S |
−1.138 |
−1.790 |
−12.676 |
−10.118 |
| 10 |
C3H4N2 |
−0.993 |
−1.255 |
−13.232 |
−10.575 |
| 11 |
C4H5N |
−1.071 |
−1.368 |
−13.868 |
−10.096 |
From Table 1 it is evident that the EA values were calculated to be more negative when using MP2 than when using B3LYP. All of these heterocycles are aromatic because of their having 6π electrons. Their aromatic behaviors were also confirmed from the negative NICS values, as shown in Table 1. We found most of the molecules to be highly σ and π-aromatic, with the exceptions including C4H4N2, C5H5N and C4H4N2. In the next section, we will discuss how we are going to make superhalogens out of these negative-electron-affinity heterocycles.
Boron-substituted aromatic heterocyclic compounds
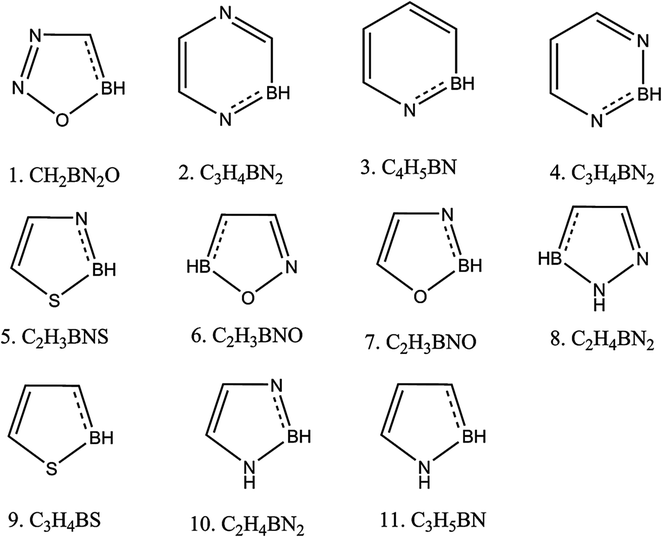
Per our strategy depicted in Scheme 1, the first step in making aromatic heterocyclic superhalogen was to form ‘B’-substituted heterocycles. So we selected the heterocycles shown in Fig. 1 and substituted one ring C atom with B. All of the systems became 5π-electronic systems when we substituted in B because boron is a trivalent atom. To regain the 6π electrons required by Huckel's rule of aromaticity, all of the systems required one extra electron. To determine whether this extra electron makes any difference in the electron affinity (EA) values, we optimized both the anionic and neutral forms of the B-substituted heterocycles. All of the optimized structures are shown in (ESI, Fig. 2†). The schematic representations of such systems are given in Fig. 2 and their EA values are tabulated in Table 2. From the table it is evident that all of the EA values became positive after boron substitution in the ring but not enough to have superhalogen properties. To have superhalogen properties, molecules should display EA values higher than that of chlorine (3.6 eV). As an example, the EA values of compound 1 (Table 1), which were calculated using B3LYP and MP2 levels of theory to be −0.333 eV and −1.277, respectively, became 1.945 eV for B3LYP and 3.328 eV for MP2 when we substituted one C of the aromatic ring with a B. Similarly, the EA of compound 4 in Table 1 changed from −0.314 eV to 2.802 eV. To determine the aromaticity of the anions of the B-substituted heterocycles, we calculated NICS(0) and NICS(1). All of these values are listed in Table 2. Here, the NICS values of the heterocycles were calculated to decrease upon carrying out the B substitution. This decrease was due to the replacement of C by B in the ring resulting in less delocalization of the π electrons. But the NICS(1) values for the B-substituted molecules were found to be sufficiently negative to consider these molecules to be π-aromatic. Therefore, although B substitution increased the EA values from negative to positive, we could consider these molecules to not be superhalogens.
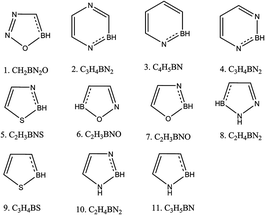 |
| | Fig. 2 Schematic diagrams of the aromatic heterocyclic molecules with each one containing one carbon substituted with a boron. | |
Table 2 Calculated EA values for boron-substituted aromatic heterocyclic systems at the B3LYP/6-31+G(d,p) and MP2/6-31+G(d,p) levels of theory. NICS(0) and NICS(1) values were computed at the B3LYP/6-31+G(d,p) level of theory
| Sl. no. |
Boron substituted molecules |
EA (eV) (B3LYP) |
EA (eV) (MP2) |
NICS(0) [ppm] (B3LYP) |
NICS(1) [ppm] (B3LYP) |
| 1 |
CH2N2BO |
1.945 |
3.328 |
−9.113 |
−10.497 |
| 2 |
C3H4BN2 |
2.895 |
3.644 |
−3.085 |
−7.628 |
| 3 |
C4H5BN |
2.521 |
3.268 |
−5.001 |
−7.862 |
| 4 |
C3H4BN2 |
2.802 |
2.615 |
−2.995 |
−6.142 |
| 5 |
C2H3BNS |
2.446 |
2.710 |
−7.953 |
−10.092 |
| 6 |
C2H3BNO |
2.602 |
2.748 |
−9.214 |
−7.596 |
| 7 |
C2H3BNO |
1.572 |
2.271 |
−7.837 |
−6.328 |
| 8 |
C2H4BN2 |
1.019 |
2.325 |
−10.858 |
−8.664 |
| 9 |
C3H4BS |
2.469 |
2.081 |
−10.244 |
−7.896 |
| 10 |
C2H4BN2 |
1.852 |
1.721 |
−10.056 |
−7.405 |
| 11 |
C3H5BN |
1.385 |
1.195 |
−7.271 |
−10.523 |
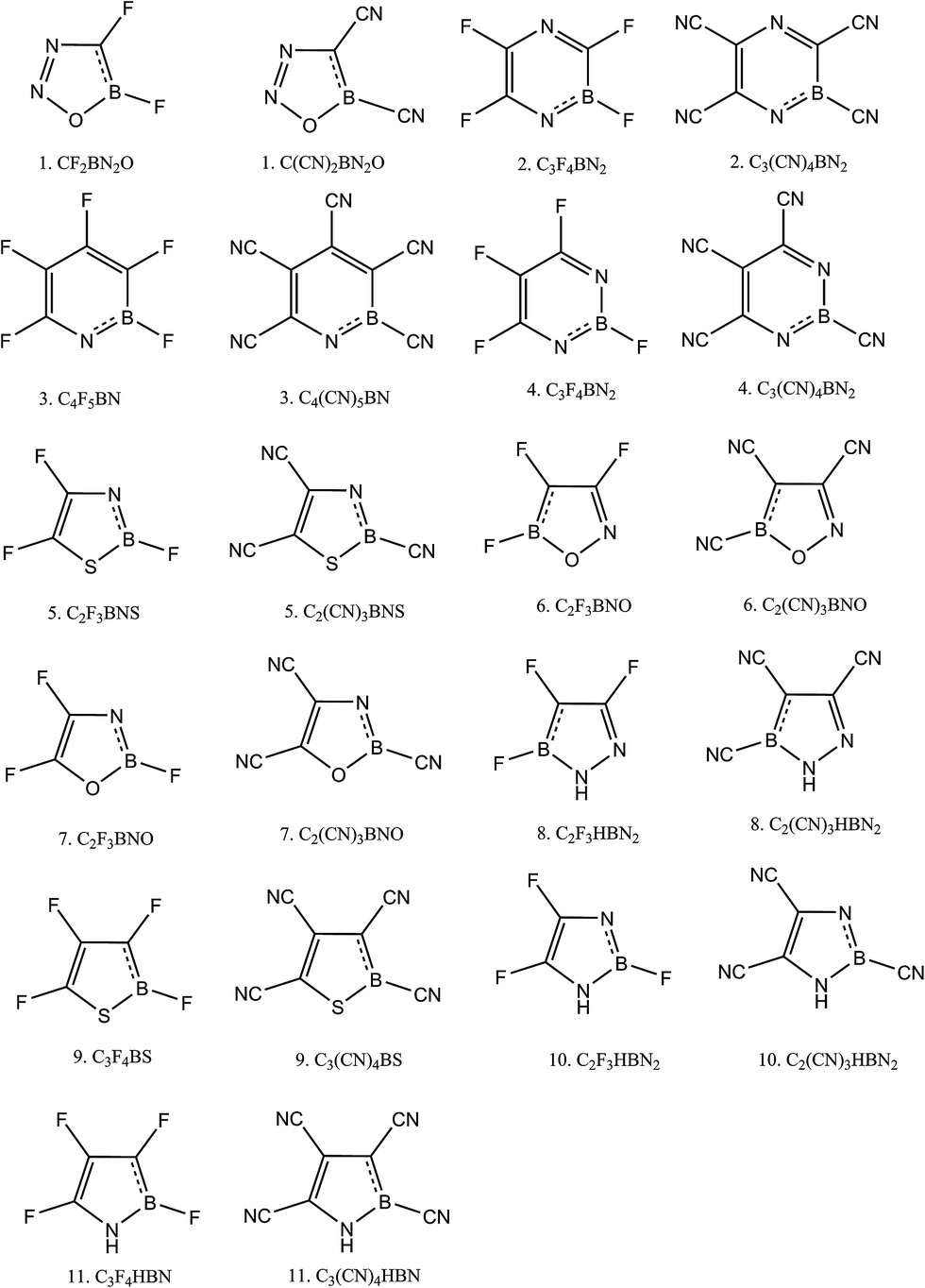
As our primary goal was to make superhalogen molecules, we proceeded to our second phase of Scheme 1. Here we substituted H by two groups: F and CN. Both F and CN are electron withdrawing in nature and have EA values of 3.4 eV and 3.86 eV, respectively. Here, instead of calculating EA, we calculated the vertical detachment energy (VDE) to determine whether the molecules are superhalogens. To calculate VDE, first we optimized the anion geometry and then calculated the energy of neutral species based on the optimized anion geometry. All of the optimized structures are depicted in (ESI, Fig. 3†). The schematic diagrams of F- and CN-substituted heterocycles are presented in Fig. 3, and their corresponding VDE and NICS(0,1) values are given in Tables 3 and 4, respectively. From Table 3 it is evident that of the F-substituted compounds, only compound 3 could be considered a superhalogen when using the B3LYP level calculation. According to the MP2 calculation, another two molecules, compounds 2 and 4, achieved superhalogen status. The rest of the molecules are not superhalogens. Regarding aromaticity, all of the molecules are aromatic. In fact, we found that π-aromaticity further decreased whereas σ-aromaticity increased upon the substitution. In contrast, all of the CN-substituted heterocycles were found to be superhalogens since, as shown in Table 4, their VDE values were calculated to be higher than 3.6 eV. In fact, we can even refer to these molecules as hyperhalogens because we used a superhalogen, CN, as the ligand. The calculations using both levels of theory showed all of the CN-substituted heterocycles to be hyperhalogenic in nature. In fact, among the eleven hyperhalogens, we found compounds 2, 3 and 4 to have VDE values close to 10 eV according to the calculations using the MP2 level. The NICS(0,1) values also suggested them to have both σ and π-aromaticity. Because of the super/hyperhalogen nature, these molecules can be used as oxidizing agents. As we have mentioned earlier in the computational details section we performed a benchmark calculation on pyridine (molecule 3, Table 1), pyrrole (molecule 11, Table 1) and their derivatives by using three different range-separated functionals: LC-BLYP, LC-wPBE and wB97XD. The corresponding values are given in Table 5.
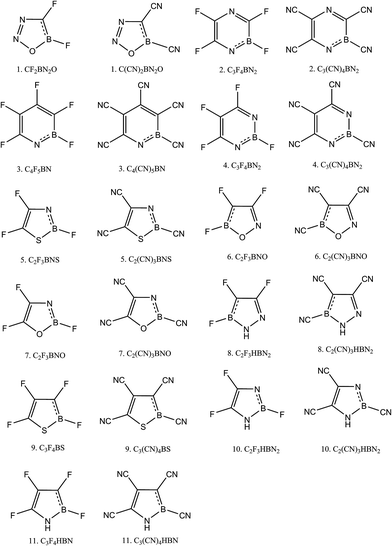 |
| | Fig. 3 Schematic diagrams of F- and CN-substituted aromatic heterocyclic compounds. | |
Table 3 Calculated VDE values for F-substituted aromatic heterocyclic systems at the B3LYP/6-31+G(d,p) and MP2/6-31+G(d,p) levels of theory. NICS(0) and NICS(1) values were computed at the B3LYP/6-31+G(d,p) level of theory
| S. no. |
F-substituted molecules |
VDE (eV) (B3LYP) |
VDE (eV) (MP2) |
NICS(0) [ppm] (B3LYP) |
NICS(1) [ppm] (B3LYP) |
| 1 |
CF2N2BO |
3.169 |
3.786 |
−9.223 |
−6.364 |
| 2 |
C3F4BN2 |
3.817 |
4.574 |
−10.825 |
−8.701 |
| 3 |
C4F5BN |
3.782 |
4.465 |
−14.261 |
−8.865 |
| 4 |
C3F4BN2 |
4.277 |
4.986 |
−9.248 |
−6.875 |
| 5 |
C2F3BNS |
3.044 |
3.403 |
−11.505 |
−5.118 |
| 6 |
C2F3BNO |
3.163 |
3.594 |
−14.389 |
−5.968 |
| 7 |
C2F3BNO |
2.788 |
3.104 |
−10.934 |
−4.096 |
| 8 |
C2HF3BN2 |
2.670 |
3.153 |
−16.915 |
−7.282 |
| 9 |
C3F4BS |
2.726 |
3.089 |
−16.912 |
−7.570 |
| 10 |
C2HF3BN2 |
2.443 |
2.816 |
−11.581 |
−4.888 |
| 11 |
C3HF4BN |
2.305 |
2.614 |
−16.406 |
−7.267 |
Table 4 Calculated VDE values for CN-substituted aromatic heterocyclic systems at the B3LYP/6-31+G(d,p) and MP2/6-31+G(d,p) level of theory. NICS(0) and NICS(1) values were computed at the B3LYP/6-31+G(d,p) level of theory
| S. no. |
CN-substituted molecules |
VDE (eV) (B3LYP) |
VDE (eV) (MP2) |
NICS(0) [ppm] (B3LYP) |
NICS(1) [ppm] (B3LYP) |
| 1 |
CCN2N2BO |
4.634 |
6.299 |
−10.971 |
−9.052 |
| 2 |
C3CN4BN2 |
5.698 |
9.379 |
−6.141 |
−8.677 |
| 3 |
C4CN5BN |
5.877 |
10.143 |
−7.504 |
−8.358 |
| 4 |
C3CN4BN2 |
5.960 |
9.516 |
−6.048 |
−8.095 |
| 5 |
C2CN3BNS |
4.794 |
7.132 |
−11.119 |
−8.719 |
| 6 |
C2CN3BNO |
4.886 |
7.126 |
−10.654 |
−8.096 |
| 7 |
C2CN3BNO |
4.676 |
6.956 |
−10.046 |
−7.738 |
| 8 |
C2HCN3BN2 |
4.475 |
6.684 |
−12.452 |
−9.055 |
| 9 |
C3CN4BS |
4.843 |
7.648 |
−12.409 |
−8.529 |
| 10 |
C2HCN3BN2 |
4.271 |
6.506 |
−11.897 |
−8.577 |
| 11 |
C3HCN4BN |
4.400 |
7.058 |
−13.527 |
−8.427 |
Table 5 Benchmark electron affinities (EA, eV) and NICS(0, ppm) values calculated for C5H5N, C4H5N and their different derivatives using different levels of theory with the 6-31+G(d,p) basis set
| Molecules |
EA (NICS 0) |
| B3LYP |
LC-BLYP |
LC-wPBE |
wB97XD |
| Values are for vertical detachment energies (VDE). |
| C5H5N |
−0.749 (−6.84) |
−0.782 (−6.75) |
−0.740 (−6.91) |
−0.854 |
| C4H5BN |
2.251 (−5.00) |
2.684 (−4.15) |
2.641 (−4.48) |
2.621 |
| C4F5BN |
3.782a (−14.26) |
3.487 (−13.96) |
3.412 (−14.01) |
3.364 |
| C4CN5BN |
5.877a (−7.504) |
6.000 (−7.87) |
5.939 (−8.09) |
5.859 |
| C4H5N |
−1.071 (−13.87) |
−1.398 (−13.44) |
−1.326 (−13.59) |
−1.390 |
| C3H5BN |
1.385 (−7.27) |
0.921 (−9.90) |
0.913 (−10.34) |
0.942 |
| C3HF4BN |
2.305a (−16.41) |
1.518 (−17.57) |
1.465 (−17.78) |
1.472 |
| C3HCN4BN |
4.400a (−13.53) |
4.349 (−13.26) |
4.319 (−13.79) |
4.300 |
As shown in Table 5, the EA and NICS(0) values were calculated to be quite similar regardless of the level used, and the CN-substituted heterocycles are indeed superhalogenic in nature.
Conclusions
In conclusion, we successfully designed superhalogens/hyperhalogens from aromatic heterocyclic compounds. The CN-substituted heterocycles we tested are hyperhalogens whereas few of the F-substituted molecules are superhalogenic in nature. The obtained nucleus-independent chemical shift values suggest all of the new heterocycles to be aromatic in nature. While aromaticity can trigger negative EA values to be switched to positive values, it is not possible to make these compounds super/hyperhalogens without suitable substituents, and hence ligand substitution also plays an important role. Increasing the electron-withdrawing nature of the ligand increases the super/hyperhalogen behavior of the molecule. We hope that these in silico results will guide the experimental synthesis of new heterocyclic molecules having an oxidizing nature.
Acknowledgements
This work is funded by the Department of Science and Technology (DST), Government of India, INSPIRE award no IFA14-CHE-151. Resourses and computational facilities of National Institute of Technology Rourkela are also greatly acknowledged.
References
- C. Kçlmel, G. Palm, R. Ahlrichs, M. Bär and A. I. Boldyrev, Chem. Phys. Lett., 1990, 173, 151 CrossRef.
- G. L. Gutsev and A. I. Boldyrev, J. Phys. Chem., 1990, 94, 2256 CrossRef CAS.
- G. L. Gutsev and A. I. Boldyrev, Chem. Phys. Lett., 1983, 101, 441 CrossRef CAS.
- S. Freza and P. Skurski, Chem. Phys. Lett., 2010, 487, 19 CrossRef CAS.
- J. Yang, X.-B. Wang, X.-P. Xing and L.-S. Wang, J. Chem. Phys., 2008, 128, 201102 CrossRef PubMed.
- G. Gutsev and A. I. Boldyrev, Chem. Phys., 1981, 56, 277 CrossRef CAS.
- S. E. Bradforth, E. H. Kim, D. W. Arnold and D. M. Neumark, J. Chem. Phys., 1993, 98, 800 CrossRef CAS.
- S. Giri, S. Behera and P. Jena, Angew. Chem., Int. Ed., 2014, 53, 13916 CrossRef CAS PubMed.
- C. Sikorska and P. Skurski, Chem. Phys. Lett., 2012, 536, 34 CrossRef CAS.
- I. Anusiewicz, J. Phys. Chem. A, 2009, 113, 6511 CrossRef CAS PubMed.
- A. I. Boldyrev and J. Simons, J. Chem. Phys., 1993, 99, 4628 CrossRef CAS.
- I. Swierszcz and I. Anusiewicz, Chem. Phys., 2011, 383, 93 CrossRef CAS.
- S. Smuczyńska and P. Skurski, Inorg. Chem., 2009, 48, 10231 CrossRef PubMed.
- G. L. Gutsev, B. K. Rao, P. Jena, X.-B. Wang and L.-S. Wang, Chem. Phys. Lett., 1999, 312, 598 CrossRef CAS.
- G. L. Gutsev, P. Jena, H.-J. Zhai and L.-S. Wang, J. Chem. Phys., 2001, 115, 7935 CrossRef CAS.
- S. Behera, D. Samanta and P. Jena, J. Phys. Chem. A, 2013, 117, 5428 CrossRef CAS PubMed.
- D. Samanta, M. M. Wu and P. Jena, Inorg. Chem., 2011, 50, 8918 CrossRef CAS PubMed.
- M. Gçtz, M. Willis, A. K. Kandalam, G. F. Gantefor and P. Jena, ChemPhysChem, 2010, 11, 853 CrossRef PubMed.
- C. Paduani and P. Jena, J. Nanopart. Res., 2012, 14, 1035 CrossRef.
- C. Sikorska and P. Skurski, Chem. Phys. Lett., 2012, 536, 34 CrossRef CAS.
- S. Giri, B. Z. Child and P. Jena, ChemPhysChem, 2014, 15, 2903 CrossRef CAS PubMed.
- H. Zhao, J. Zhou, H. Fang and P. Jena, ChemPhysChem, 2016, 17, 184 CrossRef CAS PubMed.
- K. Shen, Y. Fu, J.-N. Li, L. Liu and Q.-X. Guo, Tetrahedron, 2007, 63, 1568 CrossRef CAS.
- P. Kaur, R. Arora and N. S Gill, Indo Am. J. Pharm. Res., 2013, 3, 9067 Search PubMed.
- M. S. Saini, A. Kumar, J. Dwivedi and R. Singh, Int. J. Pharma Sci. Res., 2013, 4, 0975 Search PubMed.
- G. K. Verma, R. K. Verma and M. S. Singh, RSC Adv., 2013, 3, 245 RSC.
- E. Huckel, Z. Phys., 1931, 70, 204 CrossRef CAS.
- W. V. E. Doering and F. L. Detert, J. Am. Chem. Soc., 1951, 73, 876 CrossRef CAS.
- D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- M. Wong and T. H. Hsieh, J. Chem. Theory Comput., 2010, 6, 3704 CrossRef PubMed.
- N. Kuritz, T. Stein, R. Baer and L. Kronik, J. Chem. Theory Comput., 2011, 7, 2408 CrossRef CAS PubMed.
- M. Head-Gordon, J. A. Pople and M. J. Frisch, Chem. Phys. Lett., 1988, 153, 503 CrossRef CAS.
- H. Iikura, T. Tsuneda, T. Yanai and K. Hirao, J. Chem. Phys., 2001, 115, 3540 CrossRef CAS.
- O. A. Vydrov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 234104 CrossRef PubMed.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- P. V. R. Schleyer, H. J. Jiao, N. J. R. V. E. Hommes, V. G. Malkin and O. L. Malkina, J. Am. Chem. Soc., 1997, 119, 12669 CrossRef CAS.
- P. V. R. Schleyer, C. Maerker, A. Dransfeld, H. N. Jiao and N. J. R. V. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317 CrossRef CAS.
- P. V. R. Schleyer, M. Manoharan, Z.-X. Wang, B. Kiran, H. Jiao, R. Puchta and N. J. R. V. E. Hommes, Org. Lett., 2001, 3, 2465 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra08625j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.