
Preparation and application of novel biodegradable polyurethane copolymer
Baoli Ou*ab,
Meilong Chena,
Rao Huanga and
Hu Zhoua
aSchool of Chemistry and Chemical Engineering, Hunan University of Science and Technology, Key Laboratory of Theoretical Organic Chemistry and Function Molecule, Ministry of Education, Xiangtan 411201, People's Republic of China. E-mail: oubaoli@163.com
bState Key Laboratory of Powder Metallurgy, Central South University, Changsha 410083, People's Republic of China
First published on 28th April 2016
Abstract
Polyurethanes and polylactides are widely used due to their excellent mechanical properties, biocompatibility, and flexible construction options and their degradable, hydrolyzable and biocompatible characteristics, respectively. Herein, a novel polyurethane copolymer, poly(L-lactide)-b-(4,4′-diphenylmethane diisocyanate)-functionalized graphene (PLLA-b-PU-f-G), was prepared by the ring-opening polymerization of LLA using phenol-functionalized graphene and tin octoate as the initiator and catalyst, respectively, followed by the condensation polymerization of OH-terminated poly(L-lactide)-functionalized graphene (G-g-PLLA) and 4,4′-diphenylmethane diisocyanate (MDI). The compositional analysis and structural characterization of the resulting materials were done by FTIR, 1H NMR, TG, and DSC. The FTIR and 1H NMR results clearly showed that graphene can be covalently functionalized with polyurethane and polylactides by the proposed approach. DSC analysis indicated that with increasing graphene content, the Tg value of G-g-PLLA tends to increase mildly. A simulative ocean hanging plate experiment indicated that the copolymer material has a better antifouling performance than polyurethane. A static hydrolysis experiment showed that the incorporation of graphene increased the hydrolysis ability of polyurethane, indicating that the functionalized polyurethane can be expected to serve as a marine antifouling coating by conferring a smooth surface.
1. Introduction
Biodegradable polyurethanes (PUs) have been extensively used in various fields due to their excellent mechanical properties, biocompatibility and flexible construction options.1,2 The application areas can be expanded via functional modification. PUs with degradable performance are attractive for applications such as biomedical engineering scaffolds,3 drug delivery carriers,4 and marine antifouling coatings.5,6 Recently, the nontoxic, safe and reliable performance of PU has stimulated tremendous interest in the development of non-biocidal antifouling coatings. Jielin Ma et al.7 combined thiol–ene click reaction with condensation reaction to prepare a biodegradable polyurethane (PU) with N-(2,4,6-trichlorophenyl)maleimide pendant groups. They demonstrated that degradation can lead to a self-renewal surface, reducing the accumulation of biofouling. On the other hand, Kaibin Li8 succeeded in the synthesis of castor oil/pentaerythritol triacrylate-based UV-curable waterborne polyurethane acrylate, and a compositional and performance test demonstrated that the water resistance, glass transition temperature and thermal performance were improved.Polylactides (PLAs) are known to possess good degradation and hydrolyzation performance as well as biocompatibility,9 which provides an opportunity to further develop their applications, such as in thermoplastic plastics and films as well as fibers.10–12 Renewable, biodegradable and environmentally friendly polylactide composite materials can be formed by means of block, graft and co-polymerization. Marc A. and co-workers13 fabricated PLA-PM-PLA triblock copolymer materials via atom transfer radical polymerization (ATRP); they found that this kind of polymer possesses a low transmission temperature and an increased degradation rate. Graphene with a two-dimensional carbon nano-construction has shown significant scientific and economic application prospects in the fields of chemistry, physics, materials and electronics.14,15 In particular, the high mechanical strength and antibacterial property make it an ideal nanofiller. Recently, a number of studies indicated that the addition of graphene to composite materials can enhance the antibacterial performance of a system. Tengfei Tian et al.16 obtained GO–IONP–AgNPs composite nano-materials by growing both iron oxide nanoparticles (IONPs) and silver nanoparticles (AgNPs) on the surface of graphene oxide (GO); these nano-materials showed significantly increased antibacterial efficiency. A graphene/graphene derivative/L-lysine composite material was prepared based on electrostatic interactions and covalent bonding; this material provides potent antibacterial activity with a non-cytotoxic effect on the environment.17 Francois Perreault et al.18 described the design of thin-film composite polyamide membranes containing biocidal GO nanosheets that exhibit excellent antibacterial activity.
Recently, numerous synthetic pathways have been proposed to prepare biodegradable polyurethane materials. However, as common biodegradable polyurethane materials, polycaprolactone (PCL) and polylactic acid (PLA) usually exhibit problems such as high crystallization, slow hydrolysis rate and poor adhesion to the matrix. While considerable progress has been made in the design of biodegradable polyurethane materials, ongoing research in this area should result in the development of even better biodegradable materials in the future. In the present work, we have prepared novel polyurethane hybrid materials for which the degradation rate is controllable by adjusting the mass ratios of graphene (G) to L-lactide (LLA). A series of degradable polyurethanes were designed and synthesized using OH-terminated poly(L-lactide)-functionalized graphene, which can react with 4,4′-diphenylmethane diisocyanate (MDI), as an initiator. The covalent incorporation of graphene into a biodegradable polymer chain is expected to improve the antibacterial performance of the polymer composite material. On the other hand, the covalent chemical functionalization of graphene is favorable to prolong the service life of polyurethane materials due to its strong coordination of inorganic and organic surfaces. In addition, the incorporation of graphene into polyurethane can make the product more biodegradable. Thus, we successfully produced novel degradable polyurethane composite materials containing graphene that have high potential applications for biodegradable coatings.
2. Experimental
2.1. Materials
Natural flake graphite with an average particle size of 300 mesh and a purity of >99% was supplied by Qingdao Haida Graphite Co., Ltd, China. Dimethylformamide (DMF; 99%, Shanghai Chemical Industrial Co., Ltd., China) was purified by stirring over KOH for 48 h and then distilled under reduced pressure. L-Lactide (LLA; Aldrich) was recrystallized twice from anhydrous ethyl acetate and stored under dry nitrogen prior to use. 1,4-Butanediol (1,4-BDO; Aldrich) was dried under reduced pressure for 2 h prior to use. Toluene (Aldrich) was mixed with small pieces of sodium metal, zeolite and indicator diphenyl ketone, refluxed until the solution turned blue, and then distilled under atmospheric pressure prior to use. 3,4-Dihydroxybenzaldehyde, N-methylglycine, MDI, and stannous 2-ethylhexanoate (Sn(Oct)2) were purchased from Aldrich and used without further purification. All other reagents and solvents were purchased from commercial suppliers and used as received.2.2. Physical measurements
Fourier transform infrared (FTIR) spectra were measured with a PE Spectrum One B instrument. The spectra were collected from 64 scans with a spectral resolution of 4 cm−1 using the KBr disk method. Differential scanning calorimetry (DSC; DSC 200F3 Netzsch Co., Germany) was performed under a nitrogen flow of 50 mL min−1. After the sample was heated to 230 °C at a rate of 50 °C min−1 and equilibrated at that temperature for 5 min to remove thermal history, it was cooled to 30 °C at a rate of 10 °C min−1. It was then heated to 230 °C at a rate of 10 °C min−1. Thermogravimetric analysis (TG; NETZSCH TG 209F1) was performed under nitrogen atmosphere at a heating rate of 5 °C min−1 from room temperature to 800 °C. For the static hydrolysis experiment, a solution of 20% (w/v) polyurethane in refined tetrahydrofuran was dripped onto an epoxy resin panel (25 mm × 25 mm in size). The panels coated with PU-Gx were kept at room temperature for 5 d to completely volatilize the THF. The weight (mo) of each dried coating together with its panel was then measured before dipping into natural seawater, which was renewed once every two weeks. After 10 days, the panel was taken out, rinsed with deionized water, and dried at 60 °C in a vacuum oven for 24 h, and the weight (ms) of the panel was measured again. The mass loss was designated as (ms − mo). For the simulative ocean hanging plate experiment, we used the natural seawater. Different types of resin were dissolved in THF and then applied to epoxy plates with thicknesses of 0.5 mm. The plates were placed into the sea water to soak for 30 days followed by drying.2.3. Preparation of graphite oxide and graphene
Graphite oxide, a pseudo-two-dimensional solid in bulk form, was prepared from natural graphite powder by oxidation with KMnO4 in concentrated H2SO4 according to Hummers' method.19 Concentrated H2SO4 (46 mL) was poured into a 250 mL three-neck flask and stirred in an ice bath until the temperature dropped to 0–3 °C. Natural graphite (2.0 g) and NaNO3 (1.0 g) were added and stirred uniformly. KMnO4 (6.0 g) was gradually added with stirring and cooling in order to keep the temperature below 20 °C. The solution was heated to 35 ± 3 °C and maintained for 30 min. Distilled water was then slowly added, and the temperature was kept lower than 100 °C. After 15 min, this reaction was terminated by the addition of a large amount of distilled water and 30% H2O2 solution (5 mL). The mixture was filtered and washed with 5% HCl aqueous solution and water. The sample of graphite oxide was obtained after drying. To convert graphite oxide to GO, 100 mg graphite oxide was dispersed in 100 mL of water to create a yellow-brown dispersion, and the exfoliation of graphite oxide to GO was achieved by sonication with a cylindrical tip for 30 min. To convert GO to graphene, the resulting homogeneous solution was mixed with 80 mL of hydrazine solution as a reducing agent, and 350 mL of ammonia solution was added to adjust the pH to around 10. After stirring for a few minutes, the mixed solution was heated at about 95 °C for 1 h. The reduced product was isolated by filtration through a polytetrafluoroethylene membrane (0.22 mm), washed with water and methanol, and dried for 24 h in a vacuum oven. Then graphene (i.e., reduced GO) was obtained.2.4. Functionalization of graphene with phenol (graphene-f-OH)
To convert graphene to graphene hydroxide, 20 mg of graphene, 200 mg of 3,4-dihydroxybenzaldehyde and 200 mg of N-methylglycine were suspended in 50 mL of DMF and heated at 120 °C for 5 d. The mixture was then filtered through Millipore filters (0.45 mm) and washed thoroughly with DMF. The filtrate was sonicated in DMF for 1 h and then removed; the resulting suspension was filtered again through Millipore filters (0.45 mm). This procedure was repeated three additional times with sonication in (1) DMF, (2) 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) C2H5OH/CHCl3, and (3) diethylether. The remaining black solid was dried under vacuum (10−2 bar) for 3 d.
:1 (v/v) C2H5OH/CHCl3, and (3) diethylether. The remaining black solid was dried under vacuum (10−2 bar) for 3 d.
2.5. Synthesis of poly(L-lactic acid) grafted graphene polymer materials (G-g-PLLA)
Poly(L-lactic acid)-grafted graphene macroinitiators were synthesized as shown in Fig. 1 using a procedure similar to those described previously for the ring-opening polymerization of LLA to give PLLA-OH.20 The polymerization took place in anhydrous toluene solution at 120 °C using graphene-f-OH as the initiator and tin octoate as a catalyst. Specifically, 2 g of LLA was charged into a prepared flask, and three vacuum-nitrogen cycles were carried out. The calculated amounts of freshly prepared solutions of tin octoate (1 mL) and graphene-f-OH (0.04 g) followed by 50 mL of dry toluene were then added through a septum via a syringe under nitrogen flow. Finally, the system was closed under positive nitrogen pressure, and polymerization proceeded under stirring at 120 °C for 24 h. After reaction, the mixture was stopped by cooling to room temperature. The solidified reaction mixture was dispersed in 30 mL of methanol and then poured into 80 mL of methanol containing 1 mL of concentrated HCl to remove the Sn residue. The black solid was collected by filtration, washed with methanol, dried, re-dissolved in 10 mL of dry dichloromethane or chloroform, and precipitated dropwise into 100 mL of methanol containing 1 mL of concentrated HCl. The precipitate was collected by filtration, washed with methanol and dried in vacuo at 30 °C for 72 h.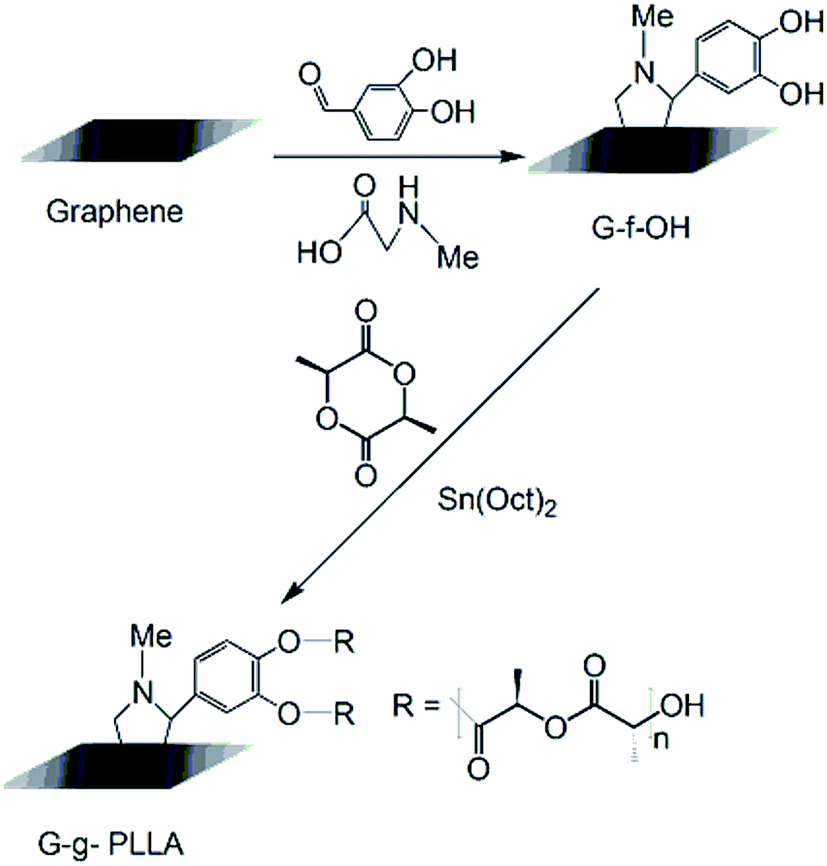 | ||
| Fig. 1 Synthesis of graphene-grafted PLLA (G-g-PLLA). | ||
2.6. Synthesis of the novel polyurethane copolymer
Polyurethane copolymer was synthesized via a condensation reaction in anhydrous toluene solution under a nitrogen atmosphere, as shown in Fig. 3. First, the reaction of 1,4-BDO (1.0 g, 0.011 mol) and 4,4′-MDI (4.0 g, 0.015 mol) was conducted at 80 °C for 2 h to yield a prepolymer, as shown in Fig. 2. Subsequently, G-g-PLLA was introduced, and the reaction was conducted at 90 °C for another 10 h. The resulting polyurethane was precipitated into excess n-hexane and dried under vacuum at 50 °C for 12 h.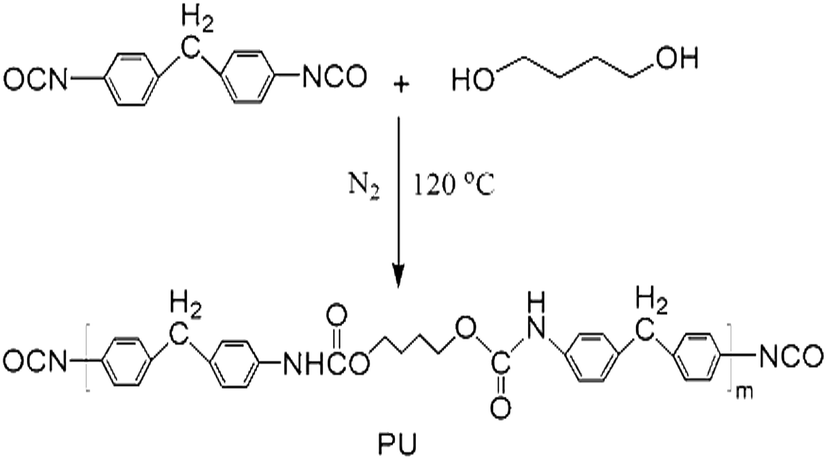 | ||
| Fig. 2 Synthesis of polyurethane pre-polymer. | ||
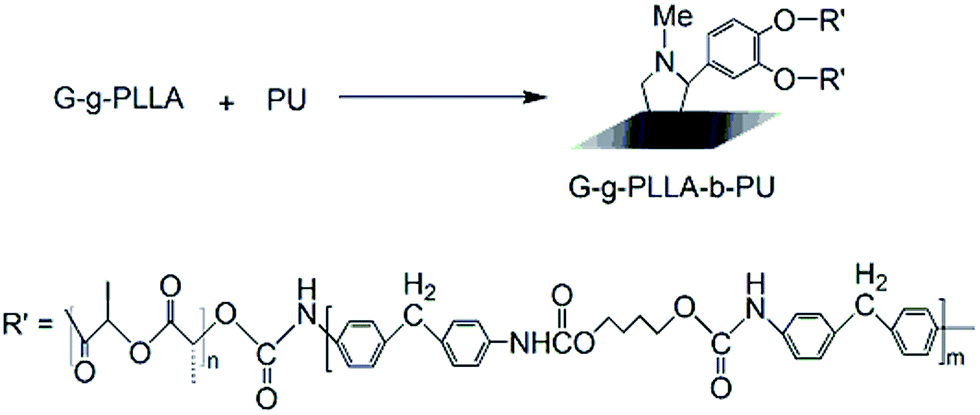 | ||
| Fig. 3 Synthesis of degradable polyurethane copolymer materials. | ||
3. Results and discussion
Our method to prepare a novel degradation polyurethane copolymer (G-g-PLLA-b-PU) consisted of three key steps, which are illustrated in Fig. 1–3.The FTIR spectra of (a) graphene and (b) graphene functionalized with phenol (G-g-OH) are shown in Fig. 4. The FTIR spectrum of graphene-f-OH shows C–H stretching features at 2950, 2922 and 2852 cm−1, which do not appear in the spectrum of pristine grapheme (curve a in Fig. 4). In addition, apparent C–C stretching bands and a weak aromatic C–H band are observed at 1100–1700 and 618 and 3040 cm−1, respectively. The C–O stretching band was observed at 1120 cm−1, and the O–H stretching band was observed at 3600–3700 cm−1, both of which are characteristic of phenols. The FTIR spectral analysis supports the successful functionalization of graphene with phenol.
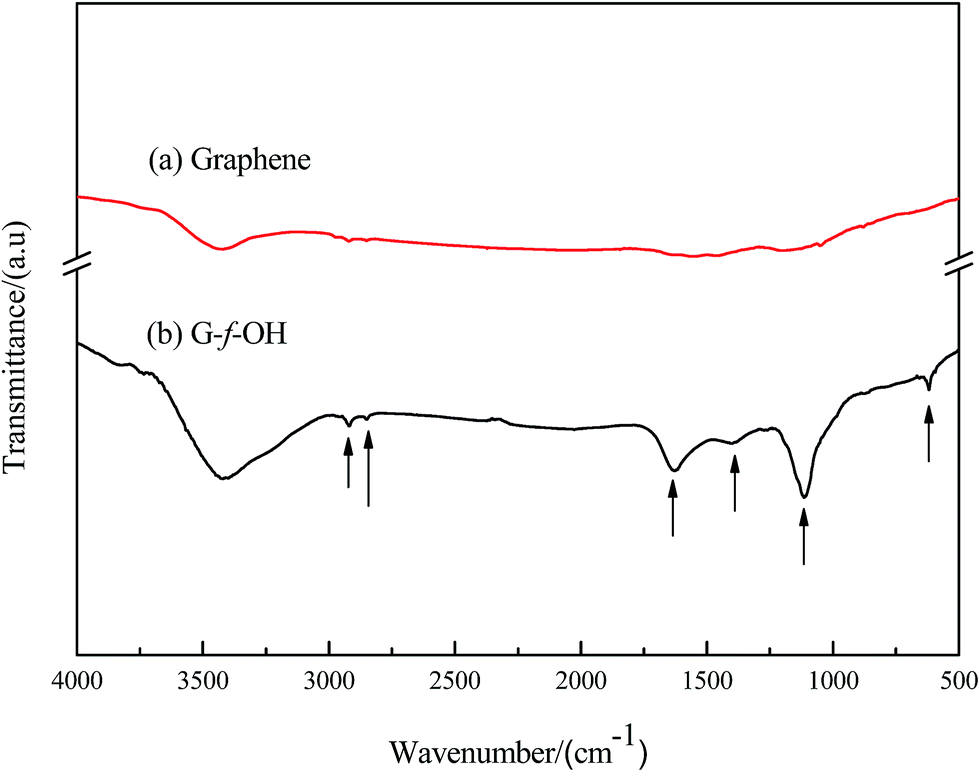 | ||
| Fig. 4 FTIR spectra of (a) graphene and (b) G-f-OH. | ||
The FTIR spectra of graphene functionalized with phenol (G-g-OH) and PLLA/g-graphene (G-g-PLLA) are shown in Fig. 5. The appearance of peaks at 1040 and 3440 cm−1 can be assigned to the stretching vibrations of C–O and –OH, respectively, which appear in the spectrum of PLLA/g-graphene. The new band at 1440 cm−1 is attributed to the bending vibration of –CH3, and the new band at 1670 cm−1 is associated with the coupling of νas(C–O–C) and δ(CH) vibration modes.21 These results clearly confirm that the L-lactide acid molecules were covalently linked to graphene, suggesting the successful surface functionalization of the graphene with OH-terminated groups.
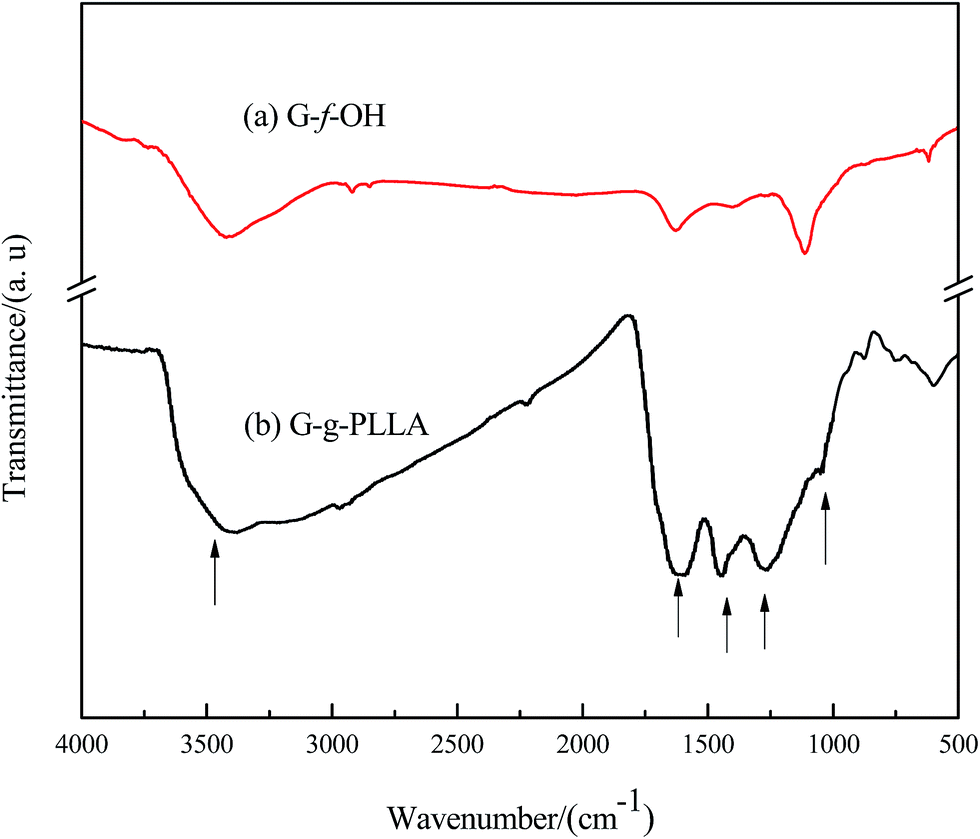 | ||
| Fig. 5 FTIR spectra of (a) G-f-OH and (b) G-g-PLLA. | ||
The FTIR spectra of PLLA/g-graphene (curve a) and the degradable polyurethane hybrid composite (G-g-PLLA-b-PU; curve b) are shown in Fig. 6. The band at 3320 cm−1 is attributable to the N–H stretching mode, and the peak at 816 cm−1 can be assigned to the bending vibration of aromatic C–H. In addition, the band at 1710 cm−1 is associated with the stretching vibration of the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in –NH–COO–, indicating the successful chemical reaction between the OH-terminated poly(L-lactide)-functionalized graphene with the NCO-terminated polyurethane prepolymer.22 The new band at 1540 cm−1 is attributed to the characteristic absorption peak of –NHCOO–. All these results indicate that the degradable polyurethane hybrid composite was successfully prepared via a combination of ring-opening polymerization and condensation polymerization.
O in –NH–COO–, indicating the successful chemical reaction between the OH-terminated poly(L-lactide)-functionalized graphene with the NCO-terminated polyurethane prepolymer.22 The new band at 1540 cm−1 is attributed to the characteristic absorption peak of –NHCOO–. All these results indicate that the degradable polyurethane hybrid composite was successfully prepared via a combination of ring-opening polymerization and condensation polymerization.
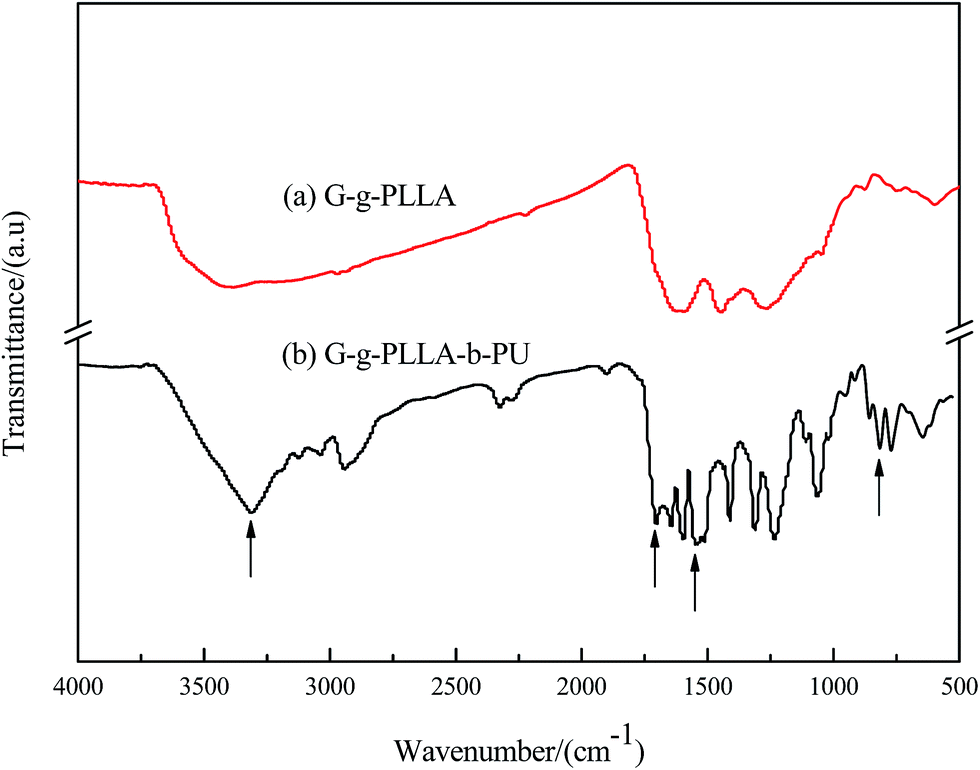 | ||
| Fig. 6 FTIR spectra of (a) G-g-PLLA and (b) G-g-PLLA-b-PU. | ||
Solution 1H NMR spectroscopy was used to further verify the successful grafting of LAA into graphene. Fig. 7 shows the typical 1H NMR spectrum of G-g-PLLA in CDCl3. As shown in Fig. 7, the chemical shifts at 5.16 and 1.53 ppm are reasonably assigned to the methylidyne proton (–CH) and methylic proton (–CH3) in the PLLA chains, respectively. The position of the c in the Fig. 7 is terminated hydroxy proton (–OH) characteristic of H absorption peak,23 position of the a in the Fig. 7 is –CH proton peak. All of these results clearly indicate that degradable PLLA was successfully grafted onto the graphene surface via ring-opening polymerization.
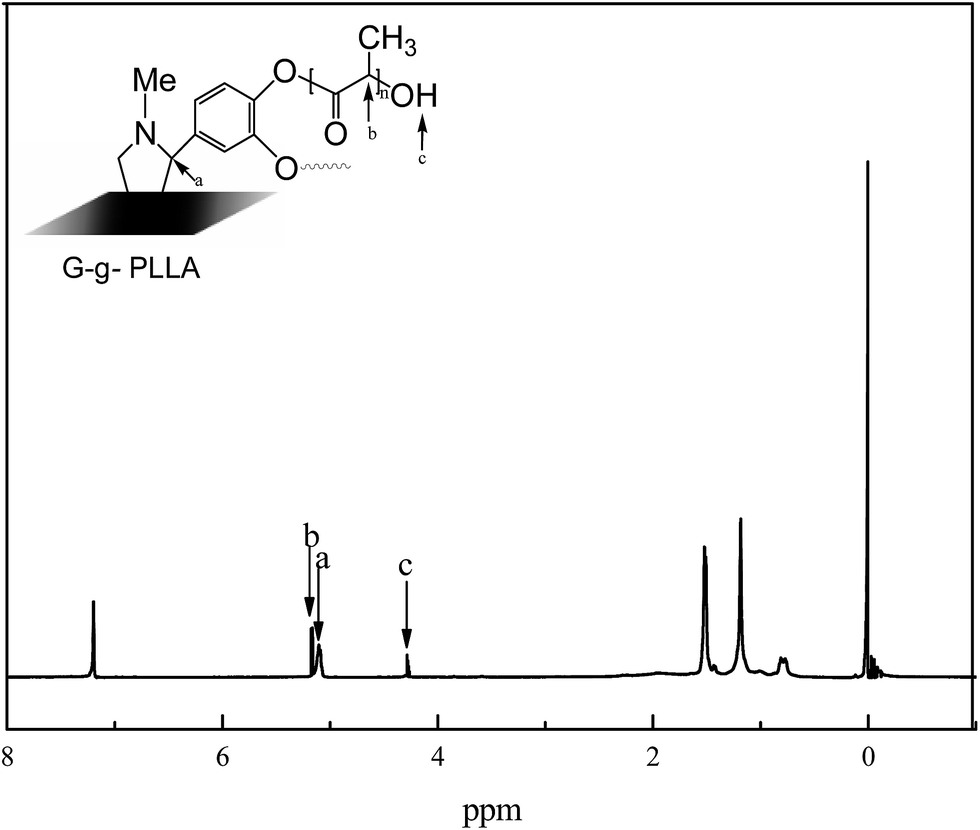 | ||
| Fig. 7 1H NMR spectrum of G-g-PLLA in CDCl3. | ||
TG thermograms of G-OH, G-g-PLLA and G-g-PLLA-b-PU polymers are shown in Fig. 8. All the materials show a little mass loss around 100 °C due to the deintercalation of H2O. The obtained G-OH sample shows a mass loss of approximately 24.6%, which is attributed to the decomposition of the grafted hydroxyl groups. Fig. 8b also shows that G-g-PLLA degraded in one step from 180 °C, and a mass loss of 43.9% occurred from 45 °C to 600 °C, which is attributed the decomposition of the grafted L-lactide. Compared with G-OH, a mass loss of 18.3% can be observed for G-g-PLLA polymer from 45 °C to 600 °C. However, as shown in Fig. 8c, the G-g-PLLA-b-PU hybrid composite degraded in two steps, the first from 80 °C to 160 °C and the second from 180 °C to 350 °C. There is a mass loss of 83.9% from 45 °C to 600 °C, which is reasonably attributed the decomposition of the grafted L-lactide and polyurethane. Compared with G-g-PLLA, a mass loss of 26% can be observed for the G-g-PLLA-b-PU copolymer composite from 45 °C to 600 °C.
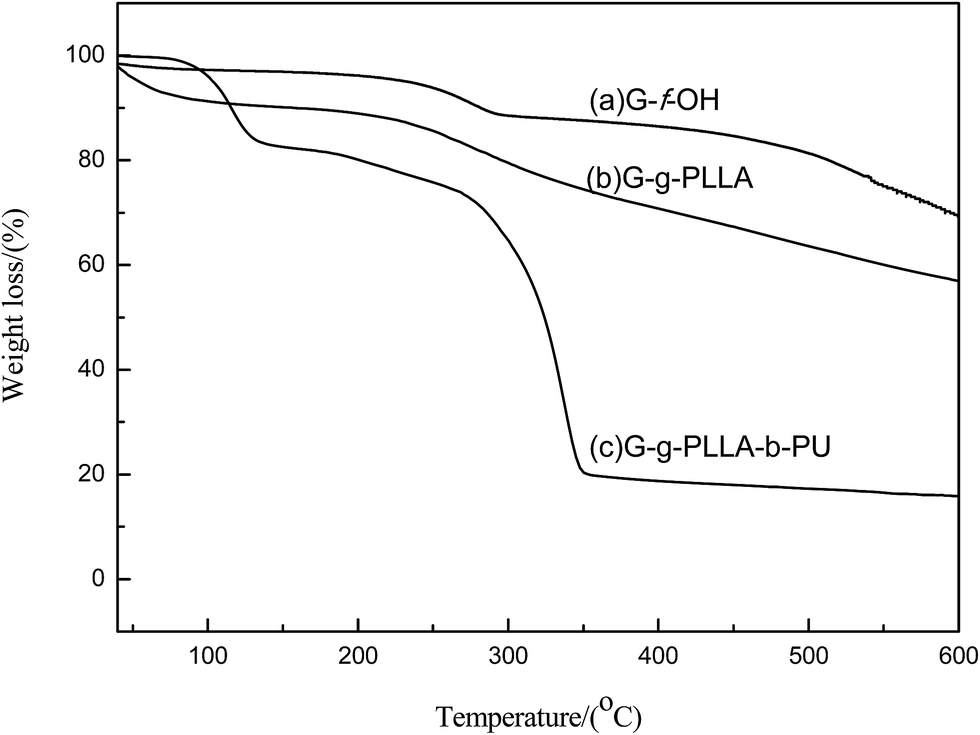 | ||
| Fig. 8 TG thermograms of (a) G-f-OH, (b) G-g-PLLA and (c) G-g-PLLA-b-PU. | ||
The DSC thermograms of the G-g-PLLA polymers are shown in Fig. 9. No crystallization peak was detected during cooling scans at scan speeds at 10 °C min−1. The DSC curves indicated that the glass transition temperature (Tg) increased from 53.7 °C to 55.2 °C as the content of graphene in creased, this phenomenon can be ascribed to the incorporation of graphene imposes constraint over the mobility of PLLA chains. A similar phenomenon in which the incorporation of inorganic nanoparticles increased the Tg values of polymers has been reported in a previous paper.24
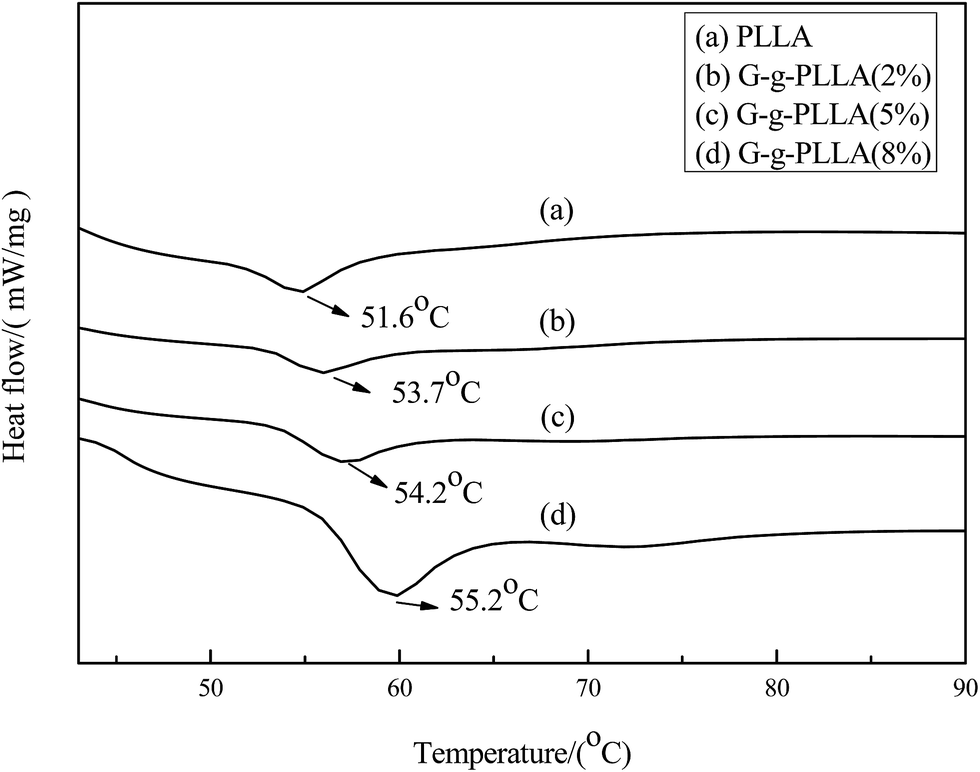 | ||
| Fig. 9 DSC curves of PU-Gx. | ||
Fig. 10 shows the mass loss of PU-Gx in natural seawater at room temperature. As can be seen from the curve, compared with PU-G0, other PU-Gx panels lost their weight more quickly, indicating that the hydrolysis rate of polyurethane increased with increasing graphene content. However, when graphene content reached 8%, the hydrolysis rate of polyurethane became slow. This phenomenon is ascribed to the disruption of PLLA crystallization caused by the introduction of graphene, resulting in increased polyurethane hydrolysis ability. Another reason may be that too much graphene limited the motion of the polymer chain, resulting in the slight increase in polyurethane hydrolysis ability.
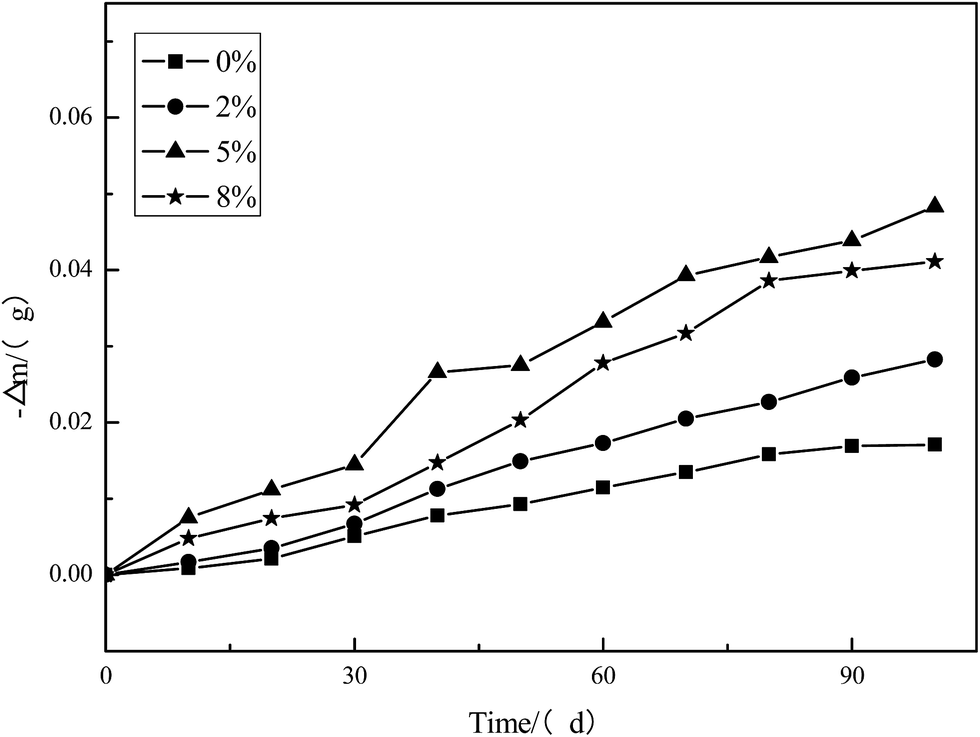 | ||
| Fig. 10 The mass loss of PU-Gx in natural seawater. | ||
Fig. 11 and 12 show the initial simulated ocean hanging plate experiment of PU and G-g-PLLA-b-PU and simulated ocean hanging plate experiment of PU and G-g-PLLA-b-PU for 30 days, respectively. During the 30 days of seawater immersion, the two resins were in the same environment, but the amount of pollutants attached was significantly different. There are many kinds of different pollutants on the surface of ordinary resin (Fig. 12a), and its surface has a small amount of degradation. In contrast, the G-g-PLLA-b-PU (Fig. 12b) surface has a small amount of contaminants attached. Compared with (b) of Fig. 11, a large amount of resin is degraded on the G-g-PLLA-b-PU ( b of Fig. 12) surface. As a result, we know that the copolymer material has better antifouling performance.
 | ||
| Fig. 11 The initial simulated ocean hanging plate experiment of PU and G-g-PLLA-b-PU. | ||
 | ||
| Fig. 12 Simulated ocean hanging plate experiment of PU and G-g-PLLA-b-PU for 30 days. | ||
4. Conclusions
A novel synthetic method for the preparation of degradable polyurethane containing graphene involving the combination of ring-opening reaction and condensation reaction was successfully demonstrated. FTIR and 1H NMR analysis indicated that PLLA can be successfully grafted onto the graphene surface. With increasing graphene content, the Tg value of the G-g-PLLA polymer tends to increase mildly because the incorporation of graphene constrains the mobility of PLLA chains. A static hydrolysis experiment indicated that the incorporation of graphene increases the hydrolysis ability of polyurethane. The G-g-PLLA-b-PU copolymer material confers a smooth surface through degradation in seawater, and the simulated ocean hanging plate experiment proved that the G-g-PLLA-b-PU copolymer material has a better antifouling performance than ordinary polyurethanes. These results indicate that functionalized PU can be expected to serve as a marine antifouling coating.Acknowledgements
This work was financially supported by the Hunan Provincial Natural Science Foundation of China (Project no. 13JJA004), Scientific Research Fund of Hunan Provincial Education Department (Project no. 15K041), Foundation of State Key Laboratory of Powder Metallurgy, and fund of National Natural Science foundation (Project no. 51443002) of People's Republic of China.References
- M. Sgarioto, R. Adhikari and P. A. Gunatillake, et al., Biomaterials, 2015, 3, 53 Search PubMed.
- Z. Ma, Y. Hong, D. M. Nelson, J. E. Pichamuthu, C. E. Leeson and W. R. Wagner, Biodegradable Polyurethane Ureas with Variable Polyester or Polycarbonate Soft Segments: Effects of Crystallinity, Molecular Weight and Composition on Mechanical Properties, Biomacromolecules, 2011, 12, 3265–3274 CrossRef CAS PubMed.
- L. Zhou, L. Yu, M. Ding, J. Li, H. Tan, Z. Wang and Q. Fu, Synthesis and Characterization of pH-Sensitive Biodegradable Polyurethane for Potential Drug Delivery Applications, Macromolecules, 2011, 44, 857–864 CrossRef CAS.
- L. Yang, J. Wei, L. Yan, Y. Huang and X. Jing, Synthesis of OH-Group-Containing, Biodegradable Polyurethane and Protein Fixation on Its Surface, J. Am. Chem. Soc., 2011, 12, 2032–2038 CAS.
- C. Ma, L. Xu, W. Xu and G. Zhang, Degradable Polyurethane for Marine Anti-biofouling, J. Mater. Chem. B, 2013, 1, 3099–3100 RSC.
- S.-H. Ye, Y. Hong and H. Sakaguchi, Nonthrombogenic, Biodegradable Elastomeric Polyurethanes with Variable Sulfobetaine Content, ACS Appl. Mater. Interfaces, 2014, 6, 22796–22797 CAS.
- J. Ma, C. Ma, Y. Yang, W. Xu and G. Zhang, Biodegradable Polyurethane Carrying Antifoulants for Inhibition of Marine Biofouling, Ind. Eng. Chem. Res., 2014, 53(32), 12753–12759 CrossRef CAS.
- K. Li, Y. Shen, G. Fei, H. Wang and J. Li, Preparation and Properties of Castor Cil/Pentaerythritol Triacrylate-based UV Curable Waterborne Polyurethane Acrylate, Prog. Org. Coat., 2015, 78, 146–154 CrossRef CAS.
- M. Brzeziński and S. Seiffert, Monodisperse Microspheres from Supramolecular Complexing Polylactides, Mater. Lett., 2015, 161, 471 CrossRef.
- K. Oksman, M. Skrifvarsb and J.-F. Selinc, Natural Fibres as Reinforcement in Polylactic Acid (PLA) Composites, Compos. Sci. Technol., 2003, 63, 1317–1324 CrossRef CAS.
- K. Gorna and S. Gogolewski, The Effect of Gamma Radiation on Molecular Stability and Mechanical Properties of Biodegradable Polyurethanes for Medical Applications, Polym. Degrad. Stab., 2003, 79, 466–474 Search PubMed.
- E. Sambha'a, A. Lallam and A. Jada, Effect of Hydrothermal Polylactic Acid Degradation on Polymer Molecular Weight and Surface Properties, J. Polym. Environ., 2010, 18, 532–538 CrossRef.
- M. A. Hillmyer and W. B. Tolman, Aliphatic Polyester Block Polymers: Renewable, Degradable, and Sustainable, Acc. Chem. Res., 2014, 47(8), 2390–2396 CrossRef CAS PubMed.
- W. Wang, S. Guo and M. Penchev, et al., Three Dimensional few Layer Graphene and Carbon Nanotube Foam Architectures for High Fidelity Supercapacitors, Nano Energy, 2013, 2, 294–303 CrossRef CAS.
- P. Nayebia and E. Zaminpayma, A Molecular Ynamic Simulation Study of Mechanical Properties of Graphene–polythiophene Composite with Reax Force Field, Phys. Lett. A, 2016, 380, 628–633 CrossRef.
- T. Tian, X. Shi, L. Cheng, Y. Luo, Z. Dong, H. Gong, L. Xu, Z. Zhong, R. Peng and Z. Liu, Graphene-Based Nanocomposite As an Effective, Multifunctional, and Recyclable Antibacterial Agent, ACS Appl. Mater. Interfaces, 2014, 6, 8542–8548 CAS.
- S. Some, S.-M. Ho, P. Dua, E. Hwang, Y. H. Shin, H. J. Yoo, J.-S. Kang, D.-ki Lee and H. Lee, Dual Functions of Highly Potent Graphene Derivative–Poly-L-Lysine Composites To Inhibit Bacteria and Support Human Cells, ACS Nano, 2012, 6(8), 7151–7161 CrossRef CAS PubMed.
- Francois Perreault, M. E. Tousley and M. Elimelech, Thin-Film Composite Polyamide Membranes Functionalized with Biocidal Graphene Oxide Nanosheets, Environ. Sci. Technol. Lett., 2014, 1(1), 71–76 CrossRef CAS.
- W. S. Hummers Jr and R. E. Offeman, Preparation of Graphitic Oxide, J. Am. Chem. Soc., 1958, 80(6), 1339 CrossRef.
- M. A. Kryuchkov, C. Detrembleur, R. Jerome, R. E. Prud'homme and C. G. Bazuin, Synthesis and Thermal Properties of Linear Amphiphilic Diblock Copolymers of L-Lactide and 2-Dimethylaminoethyl Methacrylate, Macromolecules, 2011, 44, 5209–5217 CrossRef CAS.
- J. Zhang, Y. Duan, A. J. Domb and Y. Ozaki, PLLA Mesophase and Its Phase Transition Behavior in the PLLA-PEG-PLLA Copolymer As Revealed by Infrared Spectroscopy, Macromolecules, 2010, 43, 4240–4246 CrossRef CAS.
- P. Sun, J. Wang, X. Yao, Y. Peng, X. Tu, P. Du, Z. Zheng and X. Wang, Facile Preparation of Mussel-Inspired Polyurethane Hydrogel and Its Rapid Curing Behavior, ACS Appl. Mater. Interfaces, 2014, 6, 12495–12504 CAS.
- M. A. Kryuchkov, C. Detrembleur, R. Jerome, R. E. Prud'homme and C. G. Bazuin, Synthesis and Thermal Properties of Linear Amphiphilic Diblock Copolymers of L-Lactide and 2-Dimethylaminoethyl Methacrylate, Macromolecules, 2011, 44, 5209–5217 CrossRef CAS.
- B. Ou, Z. Zhou, Q. Liu, B. Liao, S. Yi, Y. Ou, X. Zhang and D. Li, Covalent Functionalization of Graphene With Poly(methyl methacrylate) by Atom Transfer Radical Polymerization at Room Temperature, Polym. Chem., 2012, 3, 2768–2775 RSC.
| This journal is © The Royal Society of Chemistry 2016 |