
CoOx nanoparticles embedded in porous graphite carbon nanofibers derived from electrospun polyacrylonitrile@polypyrrole core–shell nanostructures for high-performance supercapacitors
Abstract
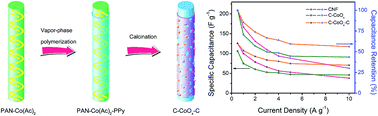
A novel composite nanostructure of C–CoOx–C with CoOx nanoparticles embedded in N-containing porous graphite carbon nanofibers (CNF) is successfully prepared via sintering the electrospun polyacrylonitrile–cobalt acetate tetrahydrate nanofibers covered by a polypyrrole (PPy) sheath that were attained from the chemical vapor-phase polymerization of pyrrole monomers using concentrated nitric acid as both the dopant and oxidant for the first time. The unique configuration with a well-defined morphology possesses a large specific surface area and prominent conductivity contributed to by the catalysis of metallic Co and the external PPy-derived carbon envelope, which could facilitate effective electron transfer and rapid ion penetration in C–CoOx–C, thus improving its electrochemical performance. As expected, when employed as an electrode active material for supercapacitors, the resultant C–CoOx–C showed a more acceptable specific capacitance, better rate capability and higher cycling stability than individual CNF and CoOx nanoparticle-decorated CNF without the coating of a N-doped carbon layer from PPy (C–CoOx).
 Please wait while we load your content...
Please wait while we load your content...

