
Magnetic molecularly imprinted polymers–silver nanoparticle based micro-solid phase extraction for the determination of polycyclic aromatic hydrocarbons in water samples
Abstract
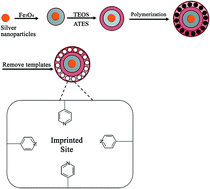
In this study, an efficient and sensitive magnetic molecularly imprinted polymer–silver nanoparticle (MMIPS) system was successfully synthesized. The MMIPS-based micro-solid phase extraction, followed by gas chromatography-flame ionization detection, was employed for the extraction and analysis of polycyclic aromatic hydrocarbons such as naphthalene, anthracene and pyrene. The adsorbent was analyzed with Fourier transform-infrared spectrometry (FT-IR) and scanning electron microscopy (SEM). The equilibrium data of the adsorbent were investigated by Scatchard analysis. In the optimized conditions, the limits of detection were achieved in the range of 0.01 to 0.09 μg L−1. Precisions were obtained from 1.1 to 6.2% for intra-day and 5.9 to 10.2% for inter-day.
 Please wait while we load your content...
Please wait while we load your content...

