
Long circulating anionic liposomes for hepatic targeted delivery of cisplatin†
Liujie Zhang,
Ying Kuang,
Jia Liu ,
Zhilan Liu*,
Shiwen Huang* and
Renxi Zhuo
,
Zhilan Liu*,
Shiwen Huang* and
Renxi Zhuo
Key Laboratory of Biomedical Polymers, Ministry of Education, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, PR China. E-mail: liuzl@whu.edu.cn; swhuang@whu.edu.cn; Tel: +86-27-68755200 Tel: +86-27-68755317
First published on 28th July 2016
Abstract
In this paper, a receptor-mediated liposomal drug delivery system (DDS) was developed aiming to deliver cisplatin (cis-diaminedichloroplatinum(II); CDDP) targeting the liver. Acetyl glycyrrhetinic acid (AGA) was chosen as the hepatic targeting ligand and acetyl glycyrrhetinic acid-poly(ethylene glycol)-stearate (AGA-PEG-ST) was synthesized. Anionic 5-cholestene-3-beta-ol-3-hemisuccinate (CHO-HS) was also synthesized. The liposomal CDDPs were prepared by employing these functional moieties with phosphatidylcholine (PC) at various ratios. Meanwhile, methoxypolyethylene glycol-stearate (MPEG-ST) with an analogous structure but without AGA was also prepared as a control. The particle sizes of AGA modified liposomes ranged from 120 nm to 180 nm and the zeta potentials were located between −39.7 mV and −3.18 mV. The liposomes had encapsulation percentages of 51.5–61.7% and a loading capacity of 23.2–26.7% for CDDP. The transmission electron microscopy (TEM) observations showed that the liposomes had spherical morphologies with homogeneous distribution. In vitro cytotoxicity of CDDP-loaded liposomes against HepG2 human liver cancer cells and A549 human lung epithelial carcinoma cells were evaluated by MTT assays. The results demonstrated that the introduction of AGA could enhance the cytotoxicity of liposomal CDDP against HepG2 cells but showed less significant impact on A549 cells. CLSM observation and FCM measurement further confirmed that AGA modified liposomes had a stronger affinity to HepG2 cells than that of liposomes without AGA. The tissue distribution of calcein in mice indicated that AGA modified liposomes resulted in higher accumulation in the liver than that of liposomes without the AGA ligand. These results demonstrated the promise of AGA decorated anionic liposomes for hepatic targeted delivery of cisplatin.
Introduction
Cancer is one of the leading causes of death in the world, while liver cancer is one of the most prevalent cancers with a high mortality.1–4 Up to now, surgery and chemotherapy are the main treatments for liver cancer, although localized primary solid tumors can be successfully removed surgically, the treatment of spreading tumors and metastases requires extensive chemotherapy.5,6 However, most anti-cancer drugs have high toxicity with poor specificity, leading to systemic toxicity and adverse effects. For instance, cisplatin, one of the most common anticancer drugs, plays an important role in the treatment of solid malignancies. Nevertheless, a number of side effects limit its application including nephrotoxicity (the major concern), neurotoxicity, ototoxicity, etc. Thus it is of great interest to develop new approaches to deliver anticancer drugs to tumorous site specifically.In order to improve therapeutic efficiency and to reduce side effects, many efforts have been devoted to develop biodegradable delivery systems with the purpose of providing higher localized dose of drug in the tumorous site.7 Among various attempts, the targeting drug-delivery system (TDDS) has shown the greatest potential.8–10
In general, TDDSs are drug carriers designed for improving the specificity of drug delivery, and they are currently employed to target desired cells, reduce the damage to the normal cells, alter biodistribution or control the release of drugs.11–13 TDDSs usually include passive targeting and active targeting.14 Passive targeting mainly leads to accumulation of drug in tumorous tissue by enhanced permeability and retention (EPR) effect.15,16 The active targeting, by contrast, is able to deliver drug to desired cells by specific recognition, thus minimizing the side effects of drugs.16,17 It is proposed that the carriers integrated passive and active targeting ability will deliver the drug to the targeted cells more efficiently.18
Many preclinical researches and clinical studies in patients revealed that multifunctional liposomes could be used as drug delivery system successfully. Conventional liposome has the intrinsic property of the passive targeting by EPR and RES,19,20 due to nanoscale size and membrane mimicking structure. However, conventional liposome easily adsorbs serum protein in the blood which causes aggregation, rapid clearance and decrease of the circulation lifetime.
In our previous work,21 long circulating anionic liposomal cisplatin was achieved by introducing poly(ethylene glycol) (PEG) into liposome, which was named “stealth liposome”.22–24 As a nontoxic polymer, PEG can shield the liposomes against destructive mechanisms in the body and improve targeting ability to tumor by EPR effect.25 In addition, in order to improve the CDDP loading efficiency of liposomes, cholesteryl hydrogen succinate (CHO-HS) was introduced to stabilize cisplatin by complex formation between CHO-HS and CDDP. The in vivo results showed that the obtained liposomes reduced the renal toxicity of platinum complexes effectively.
Based on the previous long circulating anionic liposomes, in this work, we constructed a liver-targeted DDS for cisplatin delivery by the introduction of active-targeting ligand acetyl glycyrrhetinic acid (AGA) into liposome.26 It has been reported that there are abundant receptors for GA on hepatocyte membranes.27 GA, the aglycone of glycyrrhizic acid, is one of the main bioactive compounds of licorice. What's more, it is non-toxic, cheap and easily available. Besides, GA possesses several pharmacological activities such as anti-inflammation, anti-ulcer, anti-allergenic, anti-viral and immune modulating activities,21,28–30 and it is used widely in medicine for the treatment of many pathologies.31–34
Herein, to achieve the liver-targeted delivery of cisplatin, we designed and synthesized AGA-PEG-ST, in which AGA (acetyl glycyrrhetinic acid) acts as liver targeting ligand, PEG donates the liposome stealth character, and ST helps the molecule to assemble by anchoring itself into the lipid double molecular layer. Hepatic targeting liposomes were prepared from AGA-PEG-ST, CHO-HS and PC. The performance of the AGA-PEG-ST liposomes in the hepatic targeted cellular uptake and in vivo biodistribution was evaluated.
Experimental
Materials and methods
Cholesterol (CHO) was obtained from Alfa Aesar, A Johnson Matthey Company (Shanghai, China). Cisplatin (CDDP) was purchased from Wuhan Yuancheng Gongchuang Technology Co., Ltd (Wuhan, China). Stearic acid (STA), polyethylene glycol (PEG) with molecular weight of 2000, methoxy polyethylene glycol (MPEG) with molecular weight of 1900, dimethylaminopyridine (DMAP), N,N′-dicyclohexylcarbodiimide (DCC) were purchased from Reagent Co., China. Calcein (AR) was purchased from Aladdin. All other chemicals were of analytical grade and used as received. Secondary reverse osmosis water was the laboratory homemade.Kunming mice (20 ± 5 g) were from the Laboratory Animal Centre, Wuhan University. All animals received care in compliance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals and the procedures were approved by the Wuhan University of China Animal Care and Use Committee.
Fourier transform infrared (FTIR) spectra were recorded on a Bio-Rad FTS 6000 spectrometer (Bio-Rad Company, Hercules, California, USA) using KBr pellets at room temperature. The 1H NMR spectra were obtained on VX-300 spectrometer at 300 MHz at room temperature. The samples were dissolved in trichloromethane-D (CDCl3) and tetramethylsilane (TMS) as an internal reference. The diameter and polydispersity index (PDI) of the liposomes were determined by dynamic lights scattering (DLS) with a ZETA-SIZER Nano Series ZEN3600 (Malvern Instruments Ltd., UK) at 25 °C. Each measurement was repeated three times. The physical stabilities of liposomes were also evaluated using ZEN3600. Briefly, liposomes with different compositions were stored at 4 ± 2 °C for a period of 30 days or more and assessed by DLS at proper intervals for changes in average hydrodynamic diameter. The morphologies of liposomes were observed by TEM.31 Typically, a drop of each liposome suspension was placed on copper grid with formvar film and stained with a drop of 1% phosphotungstic acid and dried. Measurement was performed with a Jeol JEM-100CXII transmission electron microscope (Tokyo, Japan) at an acceleration voltage of 100 kV.
Synthesis of methoxy polyethylene glycol-stearic acid (MPEG-ST)
STA (2.845 g, 10 mmol), DMAP (122.2 mg, 1 mmol) and DCC (2.063 g, 10 mmol) were dissolved in 30 mL anhydrous dichloromethane. The pre-dried MPEG (10 g) dissolved in 20 mL anhydrous dichloromethane was added dropwise in ice-bath. The reaction mixture was stirred overnight at room temperature. The mixture was filtered under reduced pressure and the filtrate was washed with ice water, then anhydrous magnesium sulfate was added and solution was filtered. The filtrate was concentrated and precipitated in diethyl ether. The product MPEG-ST was collected by filtration and dried under vacuum. 1H NMR (300 MHz, CDCl3): δ in ppm 3.243 (t, 2H, CH2), 0.896 (s, 3H, CH3) and 1.269 (s, 30H, 15CH2), 3.441 (m, 2H, CH2), 3.441–3.762 (m, CH2CH2O), 1.832 (s, 3H, OCH3), 4.239 (t, 2H, CH2).Synthesis of acetyl glycyrrhetinic acid-poly(ethylene glycol)-stearic acid (AGA-PEG-ST)
STA (569 mg, 2 mmol) and 3 mL thionyl chloride were dissolved in 50 mL round-bottom flask and refluxed at 60 °C for 4 h. The solvent was removed, and the product STC was obtained after re-dissolved in dichloromethane and dried for three times.GA (1.883 g, 4 mmol) and 1 mL triethylamine were dissolved in 20 mL anhydrous trichloromethane, then, 2 mL acetylchloride dissolved in 10 mL anhydrous trichloromethane was added dropwise with stirring in ice-bath. The mixture was stirred for 24 h at room temperature under nitrogen. The Et3N·HCl was filtered and the filtrate was washed with saturated brines and ice water three times in order. Then anhydrous magnesium sulfate was added and filtered, the solvent was removed under reduced pressure and the product AGA was dried under vacuum.
Following, AGA (1.025 g, 2 mmol), DCC (421.6 mg, 2 mmol) and DMAP (24.4 mg, 0.2 mmol) were dissolved in 20 mL anhydrous trichloromethane. Then, a solution of PEG 2000 (3.0 g) in 10 mL anhydrous trichloromethane was added dropwise in ice-bath. The mixture was stirred for 24 h at room temperature under nitrogen. The white solid was filtered. The filtrate was concentrated and precipitated in diethyl ether. The product AGA-PEG was collected by filtration and dried under vacuum.
AGA-PEG (2.512 g), DMAP (24.4 mg, 0.2 mmol) and 1 mL Et3N were dissolved in 30 mL anhydrous dichloromethane. STC (2 mmol) dissolved in 20 mL anhydrous dichloromethane was added dropwise with stirring in ice-bath. The reaction mixture was stirred in room temperature for 48 h. The solid was filtered and the filtrate was washed with 0.1 M HCl and ice water in order 3×, and dried with anhydrous magnesium sulfate. The filtrate was concentrated, followed by precipitation in diethyl ether. The product was collected by filtration and dried in vacuum. 1H NMR (300 MHz, CDCl3): δ in ppm 5.710 (s, 1H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 3.243 (t, 1H, CH), 2.350 (s, 2H, CH2), 2.072 (t, 1H, CH), 1.832 (t, 1H, CH), 3.441–3.765 (m, CH2CH2O), 1.253 (s, 30H, 15CH2), 4.209–4.240 (t, 2H, COCH2).
CH), 3.243 (t, 1H, CH), 2.350 (s, 2H, CH2), 2.072 (t, 1H, CH), 1.832 (t, 1H, CH), 3.441–3.765 (m, CH2CH2O), 1.253 (s, 30H, 15CH2), 4.209–4.240 (t, 2H, COCH2).
Preparation of liposomes and CDDP loaded liposomes
The mixture of phospholipids (PC, 0.05 mmol), cholesterol/cholesterol derivatives and the other additives (the relative amount compared with PC were shown in Table 1) were dissolved in 2 mL chloroform and anhydrous ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) in 250 mL eggplant shaped bottle. The solvent was evaporated under reduced pressure to obtain thin and dried lipid films. Then the films were hydrated with Tris–HCl buffer (containing 0.9% NaCl, pH 8.8) to obtain suspensions of liposomes, or hydrated with Tris–HCl buffer with 0.9% NaCl (pH 8.8) containing a certain amount of CDDP to obtain CDDP loaded liposomes. Then the liposome suspensions were extruded by a continuous high-pressure LIPEXTM Extruder (Northern Lipids Inc., Canada) using nitrogen gas for generating small unilamellar vesicles (SUV). In this process, suspensions of MLVs were passed through membrane filters with reducing pore size of 800 nm, 450 nm, 220 nm, respectively, each for ten times. The collections of extruded CDDP liposome suspensions were purified through a gel filtration column of Sephadex G-25 to remove the dissociative CDDP.
:1, v/v) in 250 mL eggplant shaped bottle. The solvent was evaporated under reduced pressure to obtain thin and dried lipid films. Then the films were hydrated with Tris–HCl buffer (containing 0.9% NaCl, pH 8.8) to obtain suspensions of liposomes, or hydrated with Tris–HCl buffer with 0.9% NaCl (pH 8.8) containing a certain amount of CDDP to obtain CDDP loaded liposomes. Then the liposome suspensions were extruded by a continuous high-pressure LIPEXTM Extruder (Northern Lipids Inc., Canada) using nitrogen gas for generating small unilamellar vesicles (SUV). In this process, suspensions of MLVs were passed through membrane filters with reducing pore size of 800 nm, 450 nm, 220 nm, respectively, each for ten times. The collections of extruded CDDP liposome suspensions were purified through a gel filtration column of Sephadex G-25 to remove the dissociative CDDP.
| No. | PC (mmol) | CHO (mmol) | CHO-HS (mmol) | MPEG-ST (mmol) | AGA-PEG-ST (mmol) |
|---|---|---|---|---|---|
| Lipo 1 | 0.05 | 0.005 | 0 | 0 | 0 |
| Lipo 2 | 0.05 | 0.005 | 0 | 0 | 0.0025 |
| Lipo 3 | 0.05 | 0.005 | 0 | 0 | 0.005 |
| Lipo 4 | 0.05 | 0.005 | 0 | 0 | 0.01 |
| Lipo 5 | 0.05 | 0 | 0.005 | 0.005 | 0 |
| Lipo 6 | 0.05 | 0 | 0.005 | 0 | 0.005 |
Encapsulation percentage and loading capacity of CDDP liposomes measured by ICP-AES
The CDDP encapsulation percentage and loading capacity were correlated with the Pt concentration in the liposomes, which was evaluated by ICP-AES after digestion with fuming nitric acid.21 Unentrapped CDDP was removed by size exclusion chromatography before measurement. Briefly, 1 mL of the CDDP liposomes, obtained from gel chromatography, was digested in 1 mL of fuming nitric acid. The mixture was heated in a vessel on hotplate placed under a hood by gradually increasing the temperature to 100 °C. The vessel was kept open to allow vigorous reaction and the evaporation of the solution to dryness (without charring). Then another 1 mL of fuming nitric acid was added to dissolve the remains and heated to a very small volume on the hotplate, till a clear solution without deposit was obtained. The digested clear liquor was then diluted with secondary reverse osmosis water to 10 mL and determined by inductively coupled plasma-atomic emission spectroscopy analysis (ICP-AES, Thermo Waltham, MA, USA). The absorbance of the solution at 157 nm was measured. The drug encapsulating percentage (EP) was calculated according to the following formula:where CE is entrapped CDDP, CT is the total amount of CDDP that added into the liposome system during the formation, while drug loading capacity (LC) was expressed by the amount of entrapped CDDP (CE) to the amount of total lipids (CL) in corresponding liposome after passing through membrane filters.

In vitro release of cisplatin
CDDP loaded liposomes were transferred to a dialysis tube (MWCO 8000–10000) respectively to measure in vitro CDDP release from liposomes.32 The liposomes 3 mL were immersed in 200 mL Tris–HCl (0.1 M, containing 0.9% NaCl, pH 7.4) and kept at 37 ± 1 °C in a water baths shaker at 60 rpm. At predetermined time intervals, 1 mL dialyzate was taken out and replaced with an equal volume of fresh buffer.33 The amount of released CDDP was analyzed by ICP-AES. The in vitro release profiles of CDDP from liposome and the accumulative release percentage of CDDP (RE%) were expressed according to experimental equation:where C0–t was the amount of CDDP released from liposome suspension from the beginning to the scheduled time, and C0 was the total amount of CDDP in liposome suspension.

In vitro cytotoxicity
The cytotoxicity of free CDDP, CDDP Lipo 5 and CDDP Lipo 6 against HepG2 cells and A549 cells were evaluated by MTT assay. Cells were seeded in a 96-well plate at a density of 6000 cells per well in 100 μL of DMEM containing 10% FBS and cultured for 24 h. Then 100 μL of DMEM medium containing CDDP or CDDP liposome was added. The cells were cultured for different time (8, 24, 48 h). The medium was removed and replaced with fresh complete DMEM. The cells were further cultured. The total cell culture time after adding drug into the cell culture medium is 48 h. And then 20 μL of MTT solution (5 mg mL−1) in PBS 7.4 was added to each well except that PBS buffer (20 μL) was added for background wells. The mixture was incubated at 37 °C for 4 h. The medium was removed and 150 μL of DMSO was added. The absorbance of the solution was measured at 570 nm using a microplate reader (Bio-Rad 550, USA). The relative cell viability was calculated according to the following equation:| Cell viability (%) = [(Asample − A0)/(Acontrol − A0)] × 100 |
Cellular uptake assays
As CDDP has no fluorescence and may potentially damage cells and influence cellular uptake, calcein with green fluorescence was selected as the model drug to investigate the liver targeting ability of AGA modified liposomes. Calcein loaded liposome (Calcein Lipo 5) and Calcein loaded AGA modified liposome (Calcein Lipo 6) was incubated with HepG2 and A549 cells respectively. After 8 h, the culture medium was removed, and washed with PBS for 3 times. The cells were fixed with 4% (w/v) paraformaldehyde aqueous solution. The nuclei of cells were stained with Hoechst 33258 (blue fluorescence). The prepared samples were observed with CLSM (Nikon, TE2000, EZ-C1, Japan).Flow cytometry test
In order to further study cellular uptake of liposomes, fluorescence intensities of calcein accumulated in HepG2 and A549 cells were examined using a flow cytometry assay. Cells were seeded in 6-well plate at a density of 400000 cells per well and incubated for 24 h. Then the cells were cultured in DMEM containing Calcein Lipo 5 or Calcein Lipo 6 at calcein concentration of 2 μg mL−1. Cells without any treatment were used as a control. After 8 h incubation, the medium was removed, and the cells were rinsed with PBS (pH 7.4) three times. Subsequently, 0.5 mL of trypsin solution was added. 1.5 min later, the cells were blowed down with DMEM containing 10% FBS and centrifuged. The cells were washed three times with PBS, and analyzed with the flow cytometer (CyAN-ADP, Beckman).
In vivo biodistribution
To investigate the tissue distribution of calcein loaded liposomes in Kunming mice, calcein loaded liposome was injected intravenously at a dose of 7 mg calcein per kg body weight. 4 h later, the mice were sacrificed. The tissues such as heart, kidney, liver, lungs and spleen were immediately removed, rinsed with saline.6,35 The fluorescence images and calcein average fluorescence intensities in heart, liver, spleen, lung and kidney were recorded by Maestro™ In-Vivo Imaging System (Cambridge Research & Instrumentation. CRI, Maestro).Results and discussion
Synthesis of MPEG-ST (A) and AGA-PEG-ST (B)
As shown in Scheme 1, methoxy polyethylene glycol-stearate (MPEG-ST) was prepared from pre-dried methoxy polyethylene glycol (MPEG) and stearic acid (STA). Acetyl glycyrrhetinic acid-poly(ethylene glycol) ester (AGA-PEG) was prepared by monoesterification of pre-dried PEG2000 with acetyl glycyrrhetinic acid (AGA). Then, AGA-PEG was further modified with STC to obtain AGA-PEG-ST. All of the products were characterized with FT-IR and 1H NMR. | ||
| Scheme 1 The synthesis of MPEG-ST (A) and AGA-PEG-ST (B). | ||
Preparation of liposomes
Phospholipids, cholesterol/cholesterol derivatives and the other additives were dissolved in the mixture of chloroform and anhydrous ethanol, and dried under reduce pressure to obtain thin and dried lipid films. Then, the films were hydrated with Tris–HCl buffer (containing 0.9% NaCl, pH 8.8) with or without CDDP to obtain corresponding liposomes suspensions. Then, the obtained liposome suspensions were extruded by a continuous high-pressure LIPEXTM Extruder, and filtered through membrane with different size (800 nm, 450 nm, 220 nm). The composition and abbreviation of various liposomes are summarized in Table 1. The stability of CDDP in Tris–HCl buffer containing 0.9% NaCl was evaluated by HPLC, the equal amount of CDDP in 0.9% NaCl was used as control. The results indicated that CDDP is stable both in Tris–HCl buffer containing 0.9% NaCl and 0.9% NaCl solution after 14 d storage at 4 °C.Liposomes sizes and zeta potentials
The mean size of liposome was measured by DLS. The average sizes of all prepared liposomes were around 100–200 nm. Fig. 1A and B show the mean sizes and polydispersity index (PDI) of liposomes without loading CDDP, Lipo 1, Lipo 2, Lipo 3, Lipo 4, Lipo 5 and Lipo 6. For Lipo 2, Lipo 3 and Lipo 4, the mean sizes decreased gradually with the increase of AGA-PEG-ST. Among the all prepared liposomes, the size of Lipo 4 was the smallest, however, the PDI of Lipo 3 was the smallest. Taking all the factors into account, Lipo 3 was chosen for the further experiments. As shown in Fig. 1C and D, compared with corresponding blank liposomes, the mean sizes of CDDP liposomes (CDDP Lipo 3, CDDP Lipo 5 and CDDP Lipo 6) were relatively smaller. Also the PDI of Lipo 5 and Lipo 6 significantly decreased after loading of CDDP. This phenomenon is consistent with the reported results in our previous study.21 It was related to the introduction of anionic CHO-HS, which interacted with cationic CDDP via complex. The relatively small sizes and uniform distribution of CDDP Lipo 5 and CDDP Lipo 6 are advantageous to their application in drug delivery.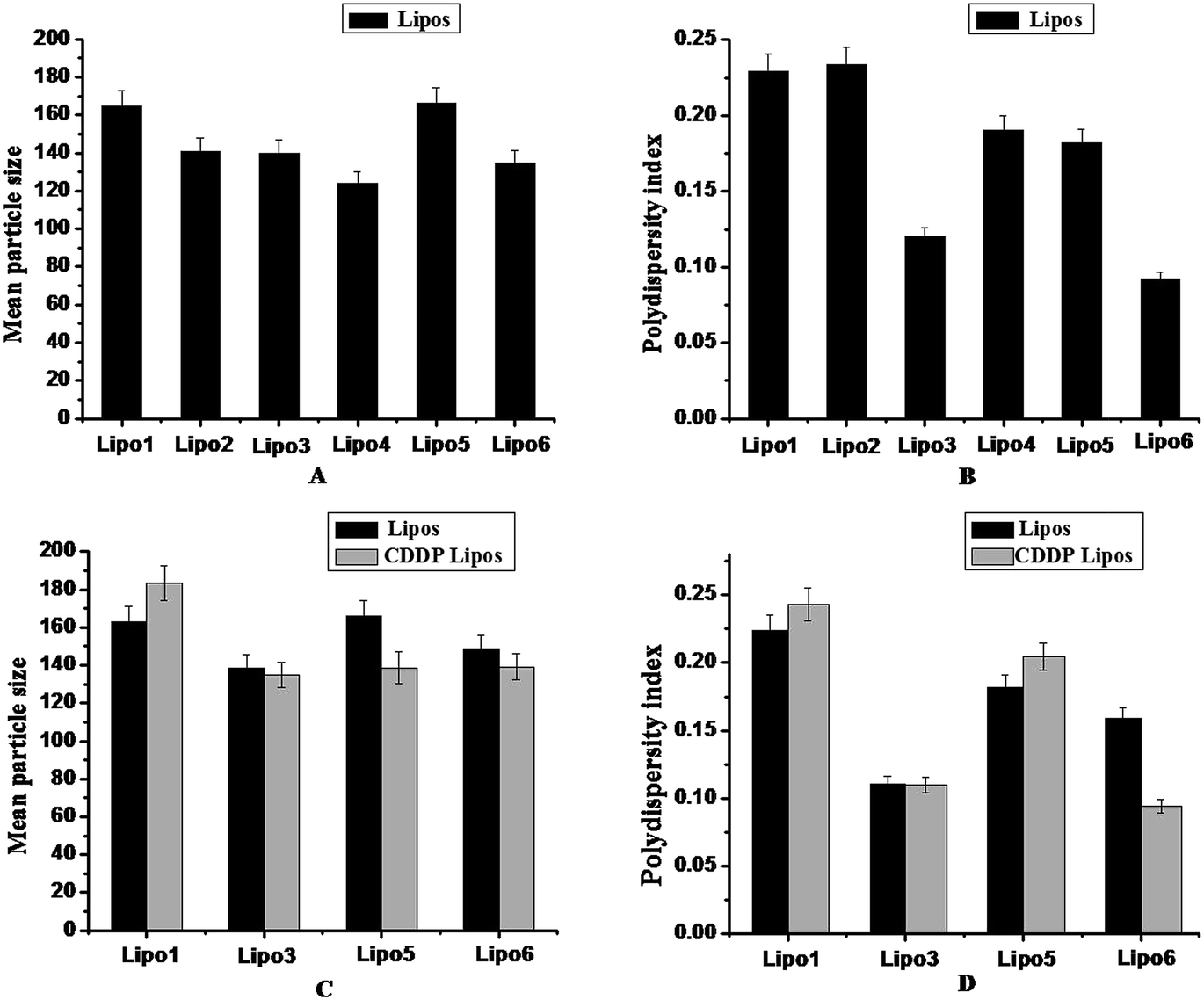 | ||
| Fig. 1 DLS results. The size of six blank Lipos (A); the size distribution index of blank Lipos (B). The size of Lipos and CDDP Lipos (C). The polydispersity index of Lipos and CDDP lipos (D). All of the Lipos were stored at 4 ± 2 °C. Each value represents the mean value and SD (n = 3). | ||
The zeta potential measurement results showed that the zeta potentials of Lipo 2 (−11.6 mV), Lipo 3 (−7.8 mV), and Lipo 4 (−3.6 mV) were slightly negative. However, the zeta potentials of Lipo 5 (−26.0 mV) and Lipo 6 (−24.7 mV) decreased significantly with the addition of negatively charged CHO-HS, which were barely associated with the AGA-PEG-ST contents. Comparison with Lipo 5 and Lipo 6, the zeta potentials of CDDP Lipo 5 (−16.3 mV) and CDDP Lipo 6 (−13.5 mV) increased after loading positively charged CDDP.
Stability of liposomes
In order to evaluate the stability of the prepared liposomes, all the liposomes were incubated in Tris–HCl buffer with 0.9% NaCl (pH 7.4) and stored at 4 °C for over 30 days. The average particle sizes and PDI of liposomes at proper incubation predetermined intervals (0, 1, 3, 7, 14, 28 and 36 days) were measured to assess their storage stability. As shown in Fig. 2A and C, only slight increase in both the particle sizes and PDI of all kinds of CDDP free liposomes were observed with the increase of incubation time. Comprehensively considering the stability and targeting ability of liposomes, Lipo 1, 3, 5 and 6 were selected for further drug loading and biological evaluation. After loading CDDP, the average diameters and PDI of CDDP Lipo 1 gradually increased, while CDDP Lipo 3, CDDP Lipo 5 and CDDP Lipo 6 were stable and showed less change in the process of storage. The high storage stabilities of CDDP Lipo 3, CDDP Lipo 5 and CDDP Lipo 6 were also attributed to the introduction of PEG segment. Besides, to evaluate the encapsulation stability of CDDP in liposomes, CDDP loaded liposomes were centrifuged at 12000 rpm, the supernatant was detected by HPLC. The results indicated that CDDP released from the liposome was less than 1% after 14 days storage at 4 °C or room temperature.
 | ||
| Fig. 2 Physical stability of Lipos and CDDP Lipos at 4 ± 2 °C storage in 36 days: the mean particle size (A) and the polydispersity index (C) of Lipos; the mean particle size (B) and the polydispersity index (D) of CDDP Lipos. Each value represents the mean value and SD (n = 3). | ||
TEM observation
The TEM images of liposomes are shown in Fig. 3. The characteristic morphologies of small unilamellar vesicles (SUV) could be observed in the TEM images of CDDP Lipo 5 and CDDP Lipo 6. The nanosized liposomes were well dispersed as individual spheres, which indicated no aggregation occurred. From the TEM images, it was also found that the diameters of CDDP Lipo 5 and CDDP Lipo 6 were around 100 nm in dried states, which were smaller than that measured by DLS analysis. | ||
| Fig. 3 Transmission electron microscopic micrograph of CDDP Lipo 5 (A) and CDDP Lipo 6 (B). | ||
Drug encapsulation percentage and loading capacity
The encapsulation percentages (EP) and loading capacities (LC) of CDDP liposomes were determined with ICP-AES, which were shown in Fig. 4. It was apparent that CDDP encapsulation efficiencies in CDDP Lipo 5 (EP 57.0% and LC 25.7%) and CDDP Lipo 6 (EP 61.7% and LC 26.7%) were higher than in conventional liposome CDDP Lipo 1 (EP 51.5% and LC 23.2%) and CDDP Lipo 3 (EP 55.7% and LC 25.1%). The higher CDDP EP and LC of Lipo 5 and Lipo 6 were due to the introduction of anionic CHO-HS, which resulted in electronic interaction between negatively charged CHO-HS and positively charged CDDP. | ||
| Fig. 4 Encapsulating percentage and loading efficiency of CDDP in CDDP Lipo 1, CDDP Lipo 3, CDDP Lipo 5 and CDDP Lipo 6. Each value represents the mean value and SD (n = 3). | ||
In vitro drug release
The release of CDDP from CDDP Lipo 1, CDDP Lipo 3, CDDP Lipo 5 and CDDP Lipo 6 were studied by the dialysis method in Tris buffer (pH 7.4) with 0.9% NaCl. The CDDP release curves from liposomes were shown in Fig. 5. All four CDDP-containing liposomes showed a rapid release of CDDP in the first 10 h and then slowed down. After 24 h, 24.3% and 23.3% of CDDP released out from CDDP Lipo 1 and CDDP Lipo 3. In the case of CDDP Lipo 5 and CDDP Lipo 6, the release of CDDP was relatively slow and total release of CDDP at 24 h was relatively low (20.7% for CDDP Lipo 5 and 19.9% for CDDP Lipo 6). The electrostatic interaction between anionic CHO-HS and cationic CDDP in CDDP Lipo 5 and CDDP Lipo 6 slowed down the release of CDDP encapsulated in the liposome. | ||
| Fig. 5 In vitro release percentage of CDDP from the CDDP Lipos to Tris–HCl buffer (pH 7.4) with 0.9% NaCl at 37 ± 1 °C on a shaker table at 60 rpm. All values are expressed as mean ± SD (n = 3). | ||
In vitro cytotoxicity
The in vitro cytotoxicities of CDDP-free liposomes and CDDP loaded liposome were evaluated with MTT assay. The effect of cellular uptake time on the cell viability was first investigated. The results, shown in Fig. 6, demonstrated that the cytotoxicities of both CDDP Lipo 5 and CDDP Lipo 6 against HepG2 cells were dependent on CDDP dose and cellular uptake time, namely, the cytotoxicity increased with increasing CDDP dose and cellular uptake time when controlling the total cell culture time at 48 h. The IC50 values of CDDP Lipo 5 against HepG2 cells were 40, 20, and 24 mg L−1 for corresponding cellular uptake time of 8 h, 24 h and 48 h, respectively. In contrast, the IC50 values of CDDP Lipo 6 against HepG2 cells were 8, 5, and 6 mg L−1 for 8 h, 24 h and 48 h cellular uptake time, respectively. For the same cellular uptake time and same total cell culture time (48 h), the cytotoxicity of CDDP Lipo 6 was significantly higher than that of CDDP Lipo 5. This means the hepatic delivery of CDDP with CDDP Lipo 6.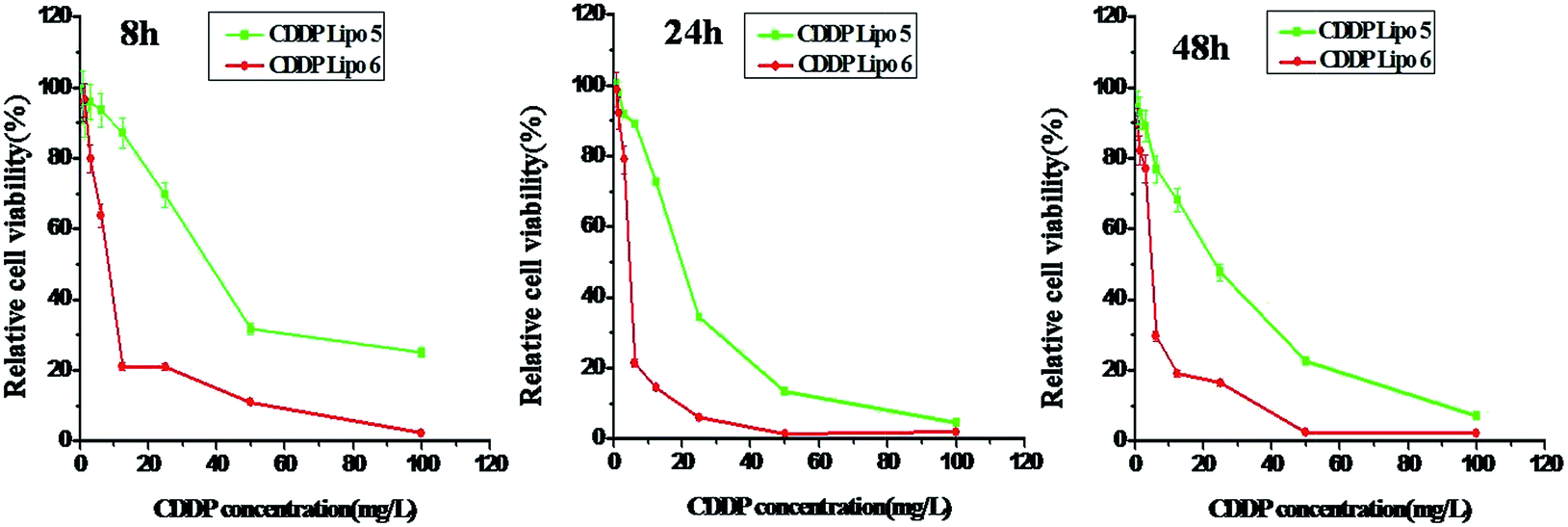 | ||
| Fig. 6 In vitro cytotoxicity assays of the CDDP Lipos to HepG2 cells after 8 h, 24 h, and 48 h incubation. Each value represents the mean value and SD (n = 4). | ||
To further investigate the hepatic specific delivery ability of CDDP Lipo 6, HepG2 cells and A549 cells were separately cultured together with CDDP Lipo 5 or CDDP Lipo 6. The results were shown in Fig. 7. Different from the case of HepG2 cells, in the case of A549 cells, both of CDDP Lipo 5 and CDDP Lipo 6 were less toxic to A549 cells, and there was no significant difference between CDDP Lipo 5 and CDDP Lipo 6. Because there was liver targeting groups AGA on the surfaces of CDDP Lipo 6, CDDP Lipo 6 showed higher toxicity to human liver cancer cells HepG2 than that of CDDP Lipo 5. However, because there were no receptors for AGA on the surfaces of A549 cells, no cytotoxicity difference could be found between CDDP Lipo 5 and CDDP Lipo 6.
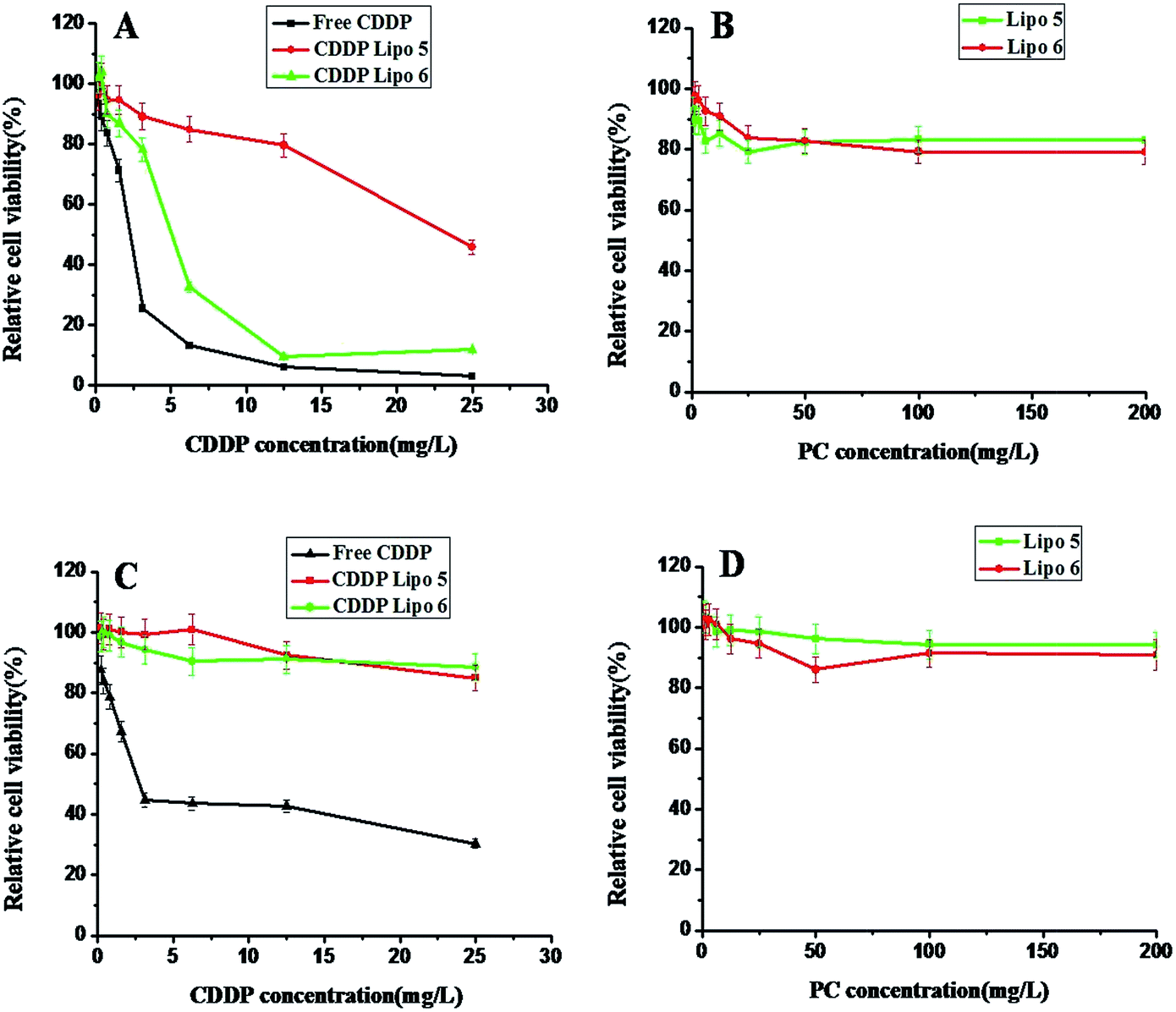 | ||
| Fig. 7 In vitro cytotoxicity assays of the liposomes with or without CDDP to HepG2 cells or A549 cells. HepG2 cells (A) and A549 cells (C) were treated with increasing concentrations of CDDP in CDDP Lipo 5 and CDDP Lipo 6, free CDDP was used as the control; HepG2 cells (B) and A549 cells (D) were treated with increasing concentrations of PC in Lipo 5 and Lipo 6. All values are expressed as mean ± SD (n = 4). | ||
The cytotoxicities of drug free liposomes, Lipo 5 and Lipo 6 were also measured. Both of Lipo 5 and Lipo 6 were almost nontoxic to HepG2 cells and A549 cells (Fig. 7B and D). This means the safety for the potential in practical applications.
Cellular uptake
To confirm the targeting ability of AGA containing liposomes to HepG2 cells, the fluorescent images of cells incubated with calcein Lipo 5 and calcein Lipo 6 were visualized by CLSM. As illustrated in Fig. 8, vivid green fluorescence could be observed clearly in HepG2 cells after 8 h incubation with calcein loaded Lipo 6 containing AGA hepatic targeting ligand (Fig. 8D), while negligible fluorescence was detected in HepG2 cells after incubation with non-hepatic targeting calcein loaded Lipo 5 (Fig. 8C). When A549 cells were similarly treated with calcein Lipo 5 and calcein Lipo 6, there was no significant difference in the intensity of green fluorescence between the cells treated with calcein Lipo 5 (Fig. 8A) and Lipo 6 (Fig. 8B). The CLSM results confirmed that the introduction of AGA into liposomes may greatly enhance the targeting ability of Lipo 6 for HepG2 cells.36 | ||
| Fig. 8 CLSM images of A549 cells incubated with calcein Lipo 5 (A) or calcein Lipo 6 (B); HepG2 cells incubated with calcein Lipo 5 (C) or calcein Lipo 6 (D), the final concentration of calcein was 2 mg L−1. Blue: Hoechst 33342; green: calcein. Scale bar is 20 μm. | ||
FCM was also used to further investigate the cellular uptake of Lipo 5 and Lipo 6. HepG2 cells and A549 cells incubated separately with calcein loaded Lipo 5 and Lipo 6 were analysed. Cells without any calcein formulation treatment were used as control and showed only auto-fluorescence.37 The mean fluorescence intensities in HepG2 cells and A549 cells after incubation with calcein Lipo 5 or calcein Lipo 6 for 8 h were illustrated in Fig. 9. The MFI of HepG2 cells treated with calcein Lipo 6 was apparently higher than that incubated with calcein Lipo 5. In the case of A549 cells, there was no significant difference between calcein Lipo 5 and calcein Lipo 6.
 | ||
| Fig. 9 Cellular uptake of liposomes detected by a flow cytometry assay. A549 cells (A) and HepG2 cells (B) incubated with calcein Lipo 5 and calcein Lipo 6 for 8 h. The blank cells were used as the control. | ||
Analyzing CLSM, FCM and cytotoxicity results, we can draw a conclusion that the AGA as a hepatic targeting ligand can hepatic specifically increase the uptake of Lipo 6 by HepG2 cells and further increase the cytotoxicity of CDDP Lipo 6 to HepG2 cells.
Biodistribution of liposomes
The in vivo biodistribution of calcein Lipo 5 and calcein Lipo 6 were visually evaluated with Maestro™ In-Vivo Imaging System. The fluorescence signal strength of calcein in mice liver and other tissues were observed after tail intravenous injection of free calcein, calcein Lipo 5 and calcein Lipo 6. As shown in Fig. 10, at 4 h after injection, there were no significant differences in the mice liver fluorescence between free calcein group and calcein Lipo 5 group. However, the liver fluorescence of hepatic targeting calcein Lipo 6 group was greatly stronger than that of free calcein group and non-targeting calcein Lipo 5 group. Quantitively analysis of fluorescence signal strength in mice liver and other tissues after injection of above three samples also showed significant differences. As shown in Fig. 11, calcein Lipo 6 accumulated extensively in the liver at 4 h after injection. The avg-signal of calcein in the liver reached 102.6 × 106 phot per cm2 per s, followed by kidney (64.4 × 106 phot per cm2 per s), lung (56.3 × 106 phot per cm2 per s), spleen (2.4 × 106 phot per cm2 per s) and heart (29.1 × 106 phot per cm2 per s) at 4 h post-injection. In contrast, calcein Lipo 5 accumulated predominantly in the mice lung, and free calcein accumulated mainly in the heart and kidney. The mean avg-signal of calcein in the liver of calcein Lipo 5 group was 41.3 × 106 phot per cm2 per s, which 2.5-fold lower than that of calcein Lipo 6 group. The results demonstrated the higher in vivo hepatic targeting ability of calcein Lipo 6 than calcein Lipo 5 and free calcein.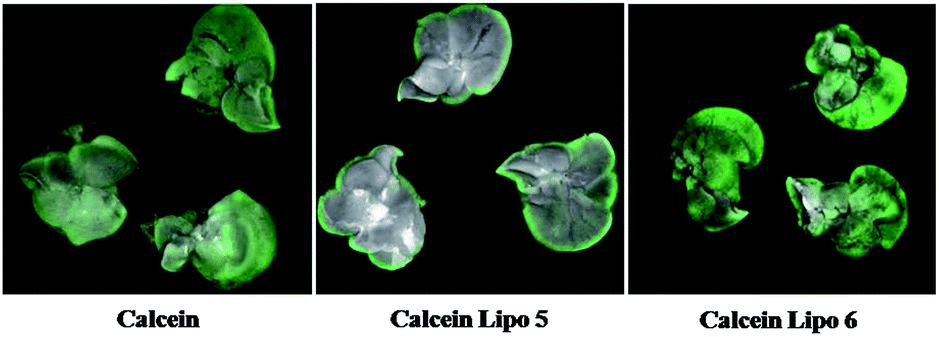 | ||
| Fig. 10 Image of mice livers after 4 h intravenous administration (n = 3). | ||
 | ||
| Fig. 11 Tissue distribution of free calcein and calcein loaded Lipos after intravenous injection for 4 h in Kunming mice at a dose of 7 mg kg−1. Each point represents average ± SD (n = 3). | ||
Conclusions
In summary, MPEG-ST, AGA-PEG-ST were successfully synthesized and separately used together with PC and CHO-HS (or CHO) to prepare a series of liposomes. The introduction of PEG segment into the liposomes resulted in the high storage stability and long circulating ability. In vitro cytotoxicity assay and in vitro cell uptake results demonstrated that AGA as a hepatic targeting group in CDDP Lipo 6 greatly increases the affinity to HepG2 cells with approximately a 4–5 fold higher in cytotoxicity against HepG2 cells in comparison to non-hepatic targeting CDDP Lipo 5. The in vivo biodistribution results also verified that AGA containing Lipo 6 accumulated particularly in the mice liver, which was different from Lipo 5 without AGA with high accumulation in the mice lung. The results suggested a potential application of AGA containing liposome as effective carriers for hepatic targeting delivery of cisplatin.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (51473127, 51273150). We would also like to thank Dr Guojun Yu for the help in the in vivo experiments.References
- Q. Tian, C. N. Zhang, X. H. Wang, W. Wang, W. Huang, R. T. Cha, C. H. Wang, Z. Yuan, M. Liu, H. Y. Wan and H. Tang, Biomaterials, 2010, 31, 4748–4756 CrossRef CAS PubMed
.
- T. Roskams, Oncogene, 2006, 25, 3818–3822 CrossRef CAS PubMed
- D. Hunecke, R. Spanel, F. Langer, S. W. Nam and J. Borlak, J. Pathol., 2012, 228, 520–533 CrossRef CAS PubMed
- R. Bansal, J. Prakash, E. Post, L. Beljaars, D. Schuppan and K. Poelstra, Hepatology, 2011, 54, 586–596 CrossRef CAS PubMed
- P. Chandna, M. Saad and Y. Wang, Mol. Pharm., 2007, 4, 11 Search PubMed
- C. Zhang, W. Wang, T. Liu, Y. Wu, H. Guo, P. Wang, Q. Tian, Y. Wang and Z. Yuan, Biomaterials, 2012, 33, 2187–2196 CrossRef CAS PubMed
- C. Jin, L. Bai, H. Wu, F. Tian and G. Guo, Biomaterials, 2007, 28, 3724–3730 CrossRef CAS PubMed
- B. Yu, S. H. Hsu, C. Zhou, X. Wang, M. C. Terp, Y. Wu, L. Teng, Y. Mao, F. Wang, W. Xue, S. T. Jacob, K. Ghoshal, R. J. Lee and L. J. Lee, Biomaterials, 2012, 33, 5924–5934 CrossRef CAS PubMed
- T. M. Allen, Nat. Rev. Cancer, 2002, 2, 750–763 CrossRef CAS PubMed
- Z. Y. He, X. Zheng, X. H. Wu, X. R. Song, G. He, W. F. Wu, S. Yu, S. J. Mao and Y. Q. Wei, Int. J. Pharm., 2010, 397, 147–154 CrossRef CAS PubMed
- W. Huang, W. Wang, P. Wang, Q. Tian, C. Zhang, C. Wang, Z. Yuan, M. Liu, H. Wan and H. Tang, Acta Biomater., 2010, 6, 3927–3935 CrossRef CAS PubMed
- T. Lammers, W. E. Hennink and G. Storm, Br. J. Cancer, 2008, 99, 392–397 CrossRef CAS PubMed
- C. Duclairoir, A. M. Orecchioni, P. Depraetere, F. Osterstock and E. Nakache, Int. J. Pharm., 2003, 253, 133–144 CrossRef CAS PubMed
- T. L. Andresen, S. S. Jensen and K. Jorgensen, Prog. Lipid Res., 2005, 44, 68–97 CrossRef CAS PubMed
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS PubMed
- J. Wu, Q. Liu and R. J. Lee, Int. J. Pharm., 2006, 316, 148–153 CrossRef CAS PubMed
- K. Maruyama, Adv. Drug Delivery Rev., 2011, 63, 161–169 CrossRef CAS PubMed
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed
- S. Aryal, C.-M. Jack Hu, V. Fu and L. Zhang, J. Mater. Chem., 2012, 22, 994–999 RSC
- J. Blaising and E. I. Pecheur, Biochimie, 2013, 95, 96–102 CrossRef CAS PubMed
- Y. Kuang, J. Liu, Z. L. Liu and R. X. Zhuo, Biomaterials, 2012, 33, 1596–1606 CrossRef CAS PubMed
- M. C. Branco and J. P. Schneider, Acta Biomater., 2009, 5, 817–831 CrossRef CAS PubMed
- Y. C. Li, S. Rissanen, M. Stepniewski, O. Cramariuc, T. Rog, S. Mirza, H. Xhaard, M. Wytrwal, M. Kepczynski and A. Bunker, J. Phys. Chem. B, 2012, 116, 7334–7341 CrossRef CAS PubMed
- K. Na, S. A. Lee, S. H. Jung, J. Hyun and B. C. Shin, Colloids Surf., B, 2012, 91, 130–136 CrossRef CAS PubMed
- D. Astruc, E. Boisselier and C. t. Ornelas, Chem. Rev., 2010, 110, 103 CrossRef PubMed
- Y. Cai, Y. Xu, H. F. Chan, X. Fang, C. He and M. Chen, Mol. Pharm., 2016, 13, 699–709 CrossRef CAS PubMed
- Q. Tian, X. H. Wang, W. Wang, C. N. Zhang, P. Wang and Z. Yuan, J. Nanomed. Nanotechnol., 2012, 8, 870–879 CrossRef CAS PubMed
- L. Zhang, J. Yao, J. Zhou, T. Wang and Q. Zhang, Int. J. Pharm., 2013, 441, 654–664 CrossRef CAS PubMed
- C. Fiore, M. Eisenhut, R. Krausse, E. Ragazzi, D. Pellati, D. Armanini and J. Bielenberg, Phytother. Res., 2008, 22, 141–148 CrossRef CAS PubMed
- F. Chen, J. Zhang, Y. He, X. Fang, Y. Wang and M. Chen, Biomater. Sci., 2016, 4, 167–182 RSC
- K. Ishihara, S. Takeda, Y. Wakui and S. Amagaya, J. Pharm. Pharmacol., 1996, 48, 902–905 CrossRef
- P. Kuang, W. Zhao, W. Su, Z. Zhang, L. Zhang, J. Liu, G. Ren, Z. Yin and X. Wang, Int. J. Cancer, 2013, 132, 1831–1841 CrossRef CAS PubMed
- Y. Liu, K. Qian, C. Y. Wang, C. H. Chen, X. Yang and K. H. Lee, Bioorg. Med. Chem. Lett., 2012, 22, 7530–7533 CrossRef CAS PubMed
- T. C. Kao, M. H. Shyu and G. C. Yen, J. Agric. Food Chem., 2010, 58, 8623–8629 CrossRef CAS PubMed
- P. E. Colombo, M. Boustta, S. Poujol, F. Pinguet, P. Rouanet, F. Bressolle and M. Vert, Eur. J. Pharm. Sci., 2007, 31, 43–52 CrossRef CAS PubMed
- E. Pérez-Herrero and A. Fernández-Medarde, Eur. J. Pharm. Biopharm., 2015, 93, 52–79 CrossRef
- V. Centis and P. Vermette, Colloids Surf., B, 2008, 65, 239–246 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: FTIR spectra and NMR spectra of polymer. See DOI: 10.1039/c6ra08566k |
| This journal is © The Royal Society of Chemistry 2016 |