
An alumina-coated, egg-shell Pd/α-Al2O3@SiC catalyst with enhanced ethylene selectivity in the selective hydrogenation of acetylene†
Huoli Zhanga,
Jianliang Cao*a,
Baojun Wub,
Wei Daib,
Zehua Chena and
Mingjie Maa
aSchool of Chemistry and Chemical Engineering, Henan Polytechnic University, Henan 454000, PR China. E-mail: caojianliang@hpu.edu.cn; Fax: +86-391-3986952; Tel: +86-391-3986952
bSinopec Beijing Research Institute of Chemical Industry, Beijing 100013, PR China
First published on 2nd June 2016
Abstract
For the selective hydrogenation of acetylene to ethylene, the selectivity of the palladium (Pd) catalyst is crucial. Pd is present as a skin distributed on the surface of Al2O3 supports and Pd active sites dispersed on the skin exhibit high activity for acetylene removal, which also inhibits over-hydrogenation of ethylene to ethane and the formation of oligomeric species on the catalyst. Therefore, it is significant to accurately control the distribution range of Pd active sites on the support by a feasible route. Herein, we demonstrate a facile approach to precisely control the Pd active-site distribution range on the support. An α-Al2O3 layer was prepared directly on the outside surface of a ring-like SiC substrate by one-step spray-coating, to obtain an egg-shell α-Al2O3@SiC support. The thickness of the α-Al2O3 layer could be freely tuned from 50 μm to 300 μm. The newly designed α-Al2O3@SiC support was used as the catalyst body in the selective hydrogenation of acetylene. As expected, the rational design of a Pd catalyst with this α-Al2O3@SiC composite material as the support has the ability to achieve higher ethylene selectivity than a conventional extrusive pellet Pd catalyst. In this work, the ethylene selectivity of the egg-shell Pd–Ag/α-Al2O3@SiC catalyst was 16% greater than that of a conventional pellet Pd–Ag/α-Al2O3 catalyst when achieving complete removal of acetylene in an ethylene-rich stream.
1. Introduction
Materials science and catalysis have been the focus of attention for the last three decades. Furthermore, the worldwide revolutionary developments in materials science have provided a driving force and begun to significantly influence developments in conventional porous catalysis materials, especially for key components of important heterogeneous catalysts and industry relevant catalysts for chemical processes.1–3 In fact, catalytic materials research is crucial and important to the petrochemical, environmental and chemistry industries, being involved in more than 85% of products for all fuels and chemicals. Therefore, novel materials research is not only offering many useful approaches, but is also applicable in many important practical fields.4–10In the current petrochemical industry, ethylene is an important chemical intermediate for the polymer industry (for example for polyethylene). However, it is inevitable that an ethylene-rich feed stream contains a trace amount of acetylene (0.5–2% by volume). Moreover, the small traces of acetylene can poison polymerization catalysts (such as Ziegler–Natta). Therefore, it is necessary to remove the acetylene impurity in an ethylene feed stream by selective hydrogenation. In general, the acetylene impurity must be reduced to less than 5 ppm.11–13 For the selective hydrogenation of acetylene, supported Pd catalysts are well-used in industrial processes. These supported Pd catalysts have attracted much attention and extensive work has been undertaken on them.14–24 There are two major issues with Pd catalysts, namely the addition of promoters and the modification of supports. A number of other metals have been intensively studied as promoters to improve the performance of the Pd catalysts, such as Ag,25–27 Ti,17,28 Ni,29 Sn,30 Au,31 Cu,32 K33 and Ga.22,34 Some metals or metal oxides such as Zn, Ti, La2O3 and Nb2O5 have been utilized to modify Al2O3 or SiO2 supports.35–38 For conventional industrial catalysts for acetylene selective hydrogenation, these methods have proved fruitful in improving the performance of Pd catalysts. However, palladium is present as a skin distributed on the surface of Al2O3 supports. Pd active sites dispersed on the skin exhibit high activity for acetylene removal, which also inhibits over-hydrogenation of ethylene to ethane and the formation of oligomeric species on the catalyst.39,40 Therefore, it is significant to investigate the effect of the Pd active-site distribution range on the catalytic performance of Pd catalysts.
Among the many catalyst supports so far reported (such as Al2O3, SiO2, ZrO2, CeO2, carbon, pumice, sepiolite, MCM-41, Y zeolite, polymers and so on), only the Al2O3 support has been utilized to develop a commercial catalyst for the selective hydrogenation of acetylene in the modern petrochemical industry.11,39,40 In order to achieve a feasible route to control precisely the Pd active-site distribution range on the support, silicon carbide (SiC) was used as a substrate to prepare α-Al2O3@SiC as the catalyst support for the selective hydrogenation of acetylene to ethylene. SiC has admirable mechanical strength, high thermal conductivity, excellent chemical inertness and high resistance towards oxidation. It is also a simple and inexpensive material compared to Al2O3. Moreover, SiC seems to be a potentially excellent functional material for various catalytic reactions, such as electrocatalytic reactions,41,42 photocatalytic reactions,43–46 Fischer–Tropsch synthesis47–49 and Pt catalysis for fuel cell applications.50,51
Recently, there has been widespread interest in the development of useful functional coating layer catalysts with enhanced catalytic performance. Y. Liu et al.52 systematically investigated a Co-based catalyst for Fischer–Tropsch synthesis. The Co-based catalyst was prepared using silicon carbide coated with TiO2 such that the thin layer of TiO2 enhanced cobalt active phase dispersion. This catalyst exhibited an excellent and stable catalytic activity for the Fischer–Tropsch synthesis with high C5+ selectivity. W. Song et al.53 reported that carbon-coated, methanol-tolerant platinum/graphene catalysts had an excellent long-term performance for oxygen reduction reactions and the carbon coating was found to be effective in protecting susceptible Pt nanoparticles from direct exposure to the corrosive environment and minimizing the negative effect of methanol crossover. B. Liu et al.54 synthesized a washcoating catalyst of cordierite honeycomb monoliths coated with vanadia–tungsta–titania mixed oxides. The coating process created a high dispersion of active species and the coated monolithic catalyst exhibited desirable catalytic performance in the selective catalytic reduction of NO with NH3. F. Ding et al.55 prepared SiO2-coated FeK/Al2O3 catalysts and the SiO2-coating improved the synthesis of hydrocarbons from CO2 hydrogenation. In addition, silica coated ZSM-5 composites exhibiting excellent catalytic performances have been reported by Y. Deng et al.56 and X. Qian et al.57
The ethylene selectivity of supported Pd catalysts is a crucial issue for the selective hydrogenation of acetylene in ethylene-rich feed stock. Although much progress has been made, it is unlikely that further great improvements in the ethylene selectivity for conventional pellet Pd/Al2O3 catalysts can be realized in a fixed-bed reactor. Therefore, it is compelling to synthesize a Pd/α-Al2O3@SiC catalyst with an alumina-coated egg-shell α-Al2O3@SiC composite material as the support. Moreover, the α-Al2O3@SiC composite material can facilitate the development of coated materials for catalysis in the optimized structures. To the best of our knowledge, no work has yet been documented on the α-Al2O3@SiC composite material as a functional support for a Pd catalyst for the selective hydrogenation of acetylene to ethylene. In this work, the α-Al2O3@SiC support was prepared by spray-coating using a newly designed alumina slurry. An α-Al2O3 layer was prepared directly on the outside surface of a ring-like SiC substrate by one-step coating, and good adhesion of the Al2O3 layer on SiC was achieved after calcination. Meanwhile, the thickness of the Al2O3 layer could be tuned from 50 μm to 300 μm. When used to form supported Pd catalysts, the SiC substrate could resist Pd precursor penetration, and therefore the Pd active sites were fully distributed on the Al2O3 layer.
2. Experimental
2.1 One-step coating of an Al2O3 layer on SiC
The coating slurry was prepared by dispersing α-Al2O3 powder (BET surface area 25–35 m2 g−1) in 6.25 wt% boehmite sol by milling the α-Al2O3 powder with a stirring ceramic ball mill (JM-5, Changsha Tencan Powder Technology Co., Ltd, China) at room temperature for 8 h. Determination by a laser particle size analyzer (Mastersizer 2000, Malvern Instruments Ltd, UK) showed that 90% of the powder diameters of the slurry were less than 4 μm, which was necessary to achieve good adhesion of the Al2O3 layer to the SiC substrate.The outer surface of the SiC substrate (a ring-like SiC, 8 mm in length and 6/3 mm in OD/ID) was coated once by using a high-efficiency spray coating machine (BG1-5, AVIC Beijing Aeronautical Manufacturing Technology Research Institute, China). The weight of the Al2O3 loading on the SiC substrate can be controlled according to the spray coating time at 120 °C. Then the α-Al2O3@SiC support was calcined at 1020 °C in air for 4 h. The thickness of the Al2O3 layer was tuned by the weight of the Al2O3 loading on the SiC substrate (Fig. 1). The adhesion strength of the Al2O3 layer was measured by an impact test in which the Pd/α-Al2O3@SiC catalyst was dropped along a 4.2 meter metal tubule, simulating the catalyst packing mode in the reactor. The weight loss was defined as follows:
 | (1) |
 | ||
| Fig. 1 Schematic illustration of the synthesis procedures for the Pd/α-Al2O3@SiC catalyst. | ||
2.2 Preparation of catalysts
A 500.0 g α-Al2O3@SiC support was put in a laboratory coating machine (rotating speed: 80 rpm), and 35 mL Pd(NO3)2 solution (Pd2+: 5 mg mL−1) was mist-sprayed onto the support using a home-made glass sprayer actuated by compressed air (pressure: 0.1 MPa). The Pd/α-Al2O3@SiC catalyst was subsequently dried at 120 °C for 8 h and calcined in air at 450 °C for 8 h.For comparison, conventional extrusive α-Alumina (α-Al2O3) pellets (average diameter 3.5 mm, BET surface area 28.4 m2 g−1) used as a support material were produced by the Sinopec Beijing Research Institute of Chemical Industry. A pellet Pd//α-Al2O3 catalyst was also prepared using the mist-spraying technique described above. Meanwhile, a Pd–Ag/α-Al2O3 catalyst and a Pd–Ag/α-Al2O3@SiC catalyst (14.1 wt% Al2O3 loading on SiC) were prepared by the same method as the Pd/α-Al2O3@SiC catalyst with Ag as a promoter. The four types of catalysts were analyzed by inductively coupled plasma mass spectrometry (ICP-MS 7500CX, Agilent) and the results are shown in Table 1.
| Sample | Pd-loading (wt%) | Ag-loading (wt%) |
|---|---|---|
| Pd/α-Al2O3@SiC | 0.033 | — |
| Pd/α-Al2O3 | 0.035 | — |
| Pd–Ag/α-Al2O3@SiC | 0.031 | 0.169 |
| Pd–Ag/α-Al2O3 | 0.035 | 0.176 |
2.3 Catalyst characterization
The catalysts were characterized by mercury porosimetry, X-ray powder diffraction (XRD), temperature programmed desorption of ammonia (NH3-TPD), transmission electron microscopy (TEM), and scanning electron microscopy (SEM). The pore size distributions of the catalysts were analyzed by mercury porosimetry (Quantachrome PoreMaster 60GT, USA). XRD analysis was performed on a Bruker AXS D8 Advance instrument (λ = 1.5406 Å; tube voltage, 40 kV; tube current, 300 mA). The NH3-TPD was performed to determine the acidic properties of the Al2O3 supports. Prior to NH3 adsorption, 0.1 g sample was pretreated in a U-tube quartz reactor at 600 °C for 60 min with a flow of He (30 mL min−1) and the sample was cooled down to 120 °C in a stream of He. After cooling, 30 mL min−1 10% NH3 in He was introduced into the reactor for 1 h. After NH3 adsorption, the gas was switched to He and purging was performed at 120 °C for 1 h and then the temperature was ramped from 120 °C to 600 °C with a heating rate of 10 °C min−1 under the flow of He (30 mL min−1). The NH3-TPD experiments were carried out using an AutoChem II 2920 instrument (Micromeritics Instruments, USA). TEM images were recorded by a Philips TECNAI 20 (200 kV) and SEM observations were performed on a FEI XL30 ESEM-FEG. BET surface area was determined by an ASAP 2020C instrument (Micromeritics Instruments, USA). Pd dispersion was measured using CO pulse chemisorption by an AutoChem II 2920 instrument (Micromeritics Instruments, USA).2.4 Catalytic test
For tests of catalytic performance, the selective hydrogenation of acetylene in an ethylene-rich stream was performed in an 8 mm (i.d.) stainless steel tube microreactor with 1 mL catalyst at atmospheric pressure. For comparison with the conventional pellet Pd–Ag/α-Al2O3 catalyst, an industrial side-line experiment for the selective hydrogenation of acetylene was carried out using a fixed-bed reactor (i.d. 14 mm) with 100 mL catalyst (reaction pressure: 2 MPa). Before the reaction, the catalyst was reduced by H2 (gas hourly space velocity (GHSV): 300 h−1) at 180 °C for 4 h. The typical feed stream consisted of 0.71% H2, 0.45% C2H2, 7.15% C2H6, and balanced C2H4. The total GHSV was 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 and the products were analyzed by gas chromatography (Agilent 7890) with a flame ionization detector (FID) and thermal conductivity detector (TCD). According to the literature, the acetylene conversion can be calculated by:22,25
000 h−1 and the products were analyzed by gas chromatography (Agilent 7890) with a flame ionization detector (FID) and thermal conductivity detector (TCD). According to the literature, the acetylene conversion can be calculated by:22,25
 | (2) |
The ethylene selectivity was calculated using moles of ethylene produced per mole of acetylene converted according to the following equation:22,35
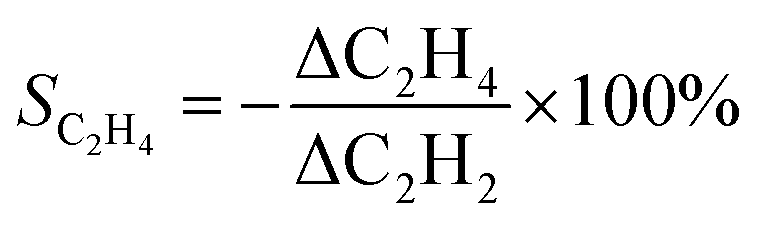 | (3) |
3. Results and discussion
In order to investigate the crystalline phases of the catalyst supports, XRD measurements were carried out. Fig. 2 shows the XRD patterns of the Al2O3 layer of the α-Al2O3@SiC support (14.1 wt% Al2O3 loading on SiC) and the conventional pellet α-Al2O3 support, which have the same characteristic α-Al2O3 peaks (2θ = 25.58, 35.15, 37.78, 43.35, 52.55, 57.5, 66.52 and 68.21, ICDD file no. 82-1468). Besides the characteristic peaks of α-Al2O3, θ-Al2O3 peaks (2θ = 31.47, 32.78, 36.68, 38.92, 44.83 and 67.42, ICDD file no. 35-0121) were also found. Fig. 2 also compares the XRD patterns of the Al2O3 layer of the α-Al2O3@SiC support before calcination and after calcination. The maximum peak of Fig. 2a is located at 2θ = 35.15 before calcination, in comparison to that for the Al2O3 layer sample after calcination and the conventional pellet α-Al2O3 support (2θ = 43.35, Fig. 2b and c). This result indicates that the boehmite sol crystal-phase transformation into α-Al2O3 or θ-Al2O3 was completed after the heat treatment.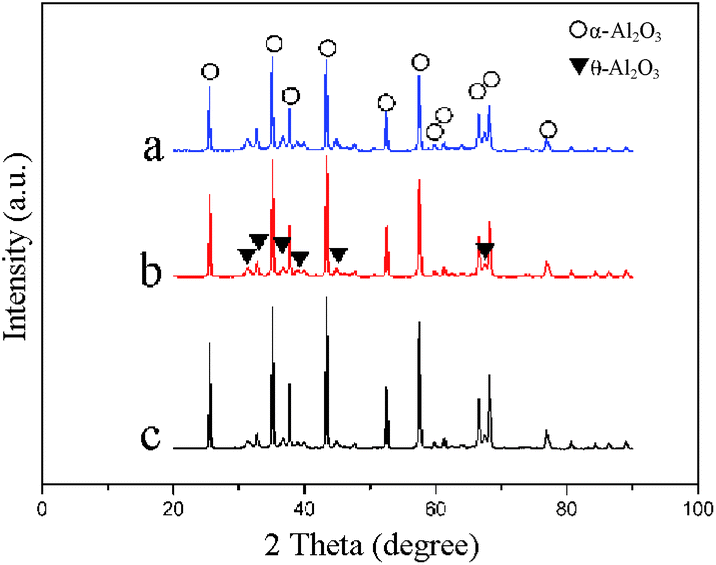 | ||
| Fig. 2 XRD patterns of (a) the Al2O3 layer of the α-Al2O3@SiC support before calcination, (b) the Al2O3 layer of the α-Al2O3@SiC support after calcination and (c) the conventional pellet α-Al2O3 support. | ||
Fig. 3 shows SEM images of the surface and cross-section morphologies of the Al2O3 layer of the α-Al2O3@SiC support (14.1 wt% Al2O3 loading on SiC) and the conventional pellet α-Al2O3 support. The surface and cross-section of the Al2O3 layer of the α-Al2O3@SiC support were smooth, homogeneous and showed dense coating before calcination (Fig. 3A and a). The particles of the Al2O3 layer are small and uniform and the particles were compact using boehmite sol as the binder. The surface and cross-section morphologies of the Al2O3 layer of the α-Al2O3@SiC support (14.1 wt% Al2O3 loading on SiC) after calcination at 1020 °C in air for 4 h are shown in Fig. 3B and b, respectively. Some conspicuous changes can be observed. It can be seen that the surface and cross-section morphologies of the Al2O3 layer were very different. The calcination resulted in the densification and shrinkage of the alumina particles. It appeared that the nanosized boehmite promoted the sintering process effectively, which is in accordance with the results reported in the literature.58,59 Fig. 3C and c show the surface and cross-section morphologies, respectively, of the conventional pellet α-Al2O3 support. It is noted that the Al2O3 support produced by the conventional extrusive process has a higher bonding strength astringing the alumina particles as compared with the Al2O3 layer produced by the one-step spray coating process. Therefore, the particles do not remain in a highly dispersed state as the surface porosity decreases.
 | ||
| Fig. 3 SEM images of the Al2O3 layer of the α-Al2O3@SiC support: (A) surface before calcination, (a) cross-section before calcination, (B) surface after calcination and (b) cross-section after calcination. SEM images of the conventional pellet α-Al2O3 support: (C) surface and (c) cross-section. | ||
Fig. 4 shows the NH3 temperature programmed desorption profiles of the Al2O3 layer of the α-Al2O3@SiC support (14.1 wt% Al2O3 loading on SiC) and the conventional pellet α-Al2O3 support. Compared with the desorption peak area of the Al2O3 layer of the α-Al2O3@SiC support before calcination, that of the Al2O3 layer of the α-Al2O3@SiC support after calcination at 1020 °C in air for 4 h was relatively low. This was due probably to the dramatic sintering process of boehmite sol because the specific surface area of the Al2O3 layer of the α-Al2O3@SiC support was decreased from 82.0 m2 g−1 to 28.0 m2 g−1 after calcination at high temperature and the quantity of total NH3 uptake was also decreased from 0.252 mmol g−1 to 0.081 mmol g−1 at the same time. For the conventional pellet α-Al2O3 support, the desorption peak area was nearly the same as that of the Al2O3 layer of the α-Al2O3@SiC support calcined at 1020 °C in air for 4 h. However, as shown in Fig. 4, all the catalyst support samples exhibited a broad peak. The NH3 desorption peak presented at 215–245 °C, which is assigned to weak acidic sites. NH3 desorption peaks corresponding to medium acidic sites and strong acidic sites were not observed. The heat treatment at high temperature reduced the acidic sites of the catalyst supports and this result is in good agreement with those reported by other researchers.60,61
 | ||
| Fig. 4 NH3-TPD profiles of (a) the Al2O3 layer of the α-Al2O3@SiC support before calcination, (b) the Al2O3 layer of the α-Al2O3@SiC support after calcination and (c) the conventional pellet α-Al2O3 support. | ||
For the one-step spray coating process, the composition of the slurry suspension is important for preparing a crack-free Al2O3 layer on the outside of the ring-like SiC substrate. In this work, α-Al2O3 powder (BET surface area 25–35 m2 g−1) was added into 6.25 wt% boehmite sol and the powder was milled with a stirring ceramic ball mill for 8 h. The distribution of particle sizes of the milled α-Al2O3 powder was measured by a laser particle size analyzer. Some 90% powder diameters of the slurry suspension were less than 4 μm, which was very effective in preventing crack formation and achieving good adhesion of the Al2O3 layer on the SiC substrate.
As shown in Fig. 5a, the Pd/α-Al2O3@SiC catalyst was prepared with 14.1 wt% Al2O3 loading on the ring-like SiC substrate and the thickness of the Al2O3 layer on the outside of the substrate was 190 ± 10 μm (Fig. S1, ESI†). The successive uniform Al2O3 layer can be observed on the outside of the ring-like SiC substrate. It also can be noted that no cracks occurred in the Al2O3 layer. The TEM image (Fig. 5b) showed that the Pd particles of the Pd/α-Al2O3@SiC catalyst were uniformly dispersed on the Al2O3 layer with diameters ranging from 4 to 10 nm. Only a few aggregations were observed; the value of Pd dispersion was 23.5%. In fact, the conventional extrusive pellet α-Al2O3 support is not generally desirable because the narrow, long pores of the pellet α-Al2O3 support could invite Pd precursor penetration. Although at least 80% Pd active sites were distributed on the surface of the pellet Pd/α-Al2O3 catalyst, the depth of Pd precursor penetration was still in the range of 1–1000 μm (Fig. 5c). The size of the Pd particles on the pellet Pd/α-Al2O3 catalyst was less than 10 nm (Fig. 5d) and the value of Pd dispersion was 19.2%. However, the depth of Pd active sites dispersed on the pellet α-Al2O3 support was more than 300 μm which is undesirable for the selective hydrogenation of acetylene to ethylene. Based on the literature, Pd active sites are present as a skin distributed on the surface of Al2O3 pellet supports which exhibit high activity for acetylene removal, and inhibit over-hydrogenation of ethylene to ethane and the formation of oligomeric species on the catalyst.39,40 Therefore, it is very significant to control the Pd active site distribution range by hindering Pd precursor penetration using the alumina-coated egg-shell α-Al2O3@SiC composite material as a support.
 | ||
| Fig. 5 (a) SEM image of the Pd/α-Al2O3@SiC catalyst, (b) TEM image of Pd particles on the Al2O3 layer of the Pd/α-Al2O3@SiC catalyst, (c) SEM image and EDS analysis of the pellet Pd/α-Al2O3 catalyst and (d) TEM image of Pd particles on the Al2O3 support of the pellet Pd/α-Al2O3 catalyst. | ||
On the other hand, α-Al2O3 is commonly used as a support for the selective hydrogenation of acetylene due to its suitable macroporous nature. In addition, the surface area of α-Al2O3 is smaller than that of γ-Al2O3 and the α-Al2O3 can provide lower dispersion of Pd active sites. According to the literature, it is desirable for the selective hydrogenation of acetylene that the Pd/α-Al2O3 catalyst possesses fewer Pd active sites for direct ethane formation than the Pd/γ-Al2O3 catalyst, which has higher ethylene selectivity62 and it was reported that α-Al2O3 was the proper material to be used as the support due to its sufficiently large pore size.39,62 Herein, mercury porosimetry was employed to determine the pore size distribution of the SiC substrate, α-Al2O3@SiC support (14.1 wt% Al2O3 loading on SiC) and conventional pellet α-Al2O3 support (Fig. 6).
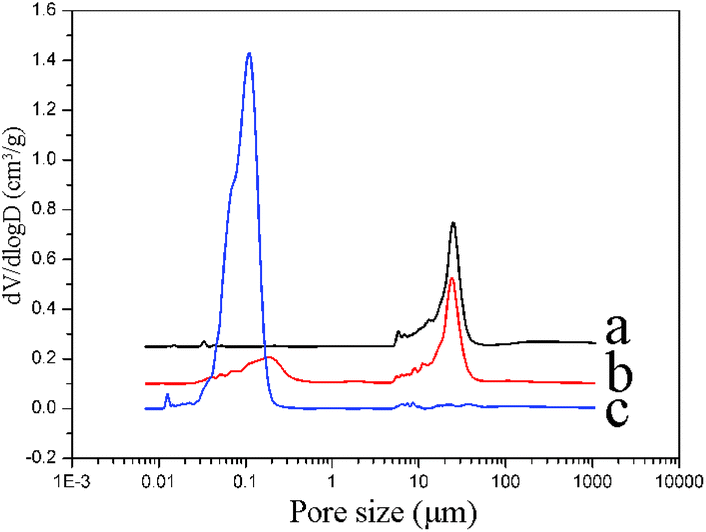 | ||
| Fig. 6 Pore size distribution curves for the samples: (a) SiC substrate, (b) α-Al2O3@SiC support and (c) conventional pellet α-Al2O3 support. The dV/dlogD value was shifted by 0.25 and 0.10 for the curves of data sets (a) and (b), respectively. | ||
As shown in Fig. 6, the Al2O3 layer of the α-Al2O3@SiC support has a similar pore size distribution to the conventional extrusive pellet α-Al2O3 support, but the SiC substrate has no such pores in the range of 30–300 nm. Moreover, for the Al2O3 layer on SiC substrate, the main pore size distribution is in the range of 100 nm to 300 nm. Meanwhile, the pore size distribution is broadened compared with the conventional extrusive pellet α-Al2O3 support. The largest pore sizes of the α-Al2O3@SiC support are increased from 200 nm to 300 nm. The increase of the pore size of α-Al2O3@SiC support might offer a benefit for the selective hydrogenation of acetylene to ethylene.
Developing a facile approach to precisely control the Pd active-site distribution range on Al2O3 supports is of significance for supported Pd catalysts. Herein, the α-Al2O3 layer was prepared directly on the outside surface of the ring-like SiC substrate by one-step spray-coating, which resulted in an egg-shell α-Al2O3@SiC support. The thickness of the α-Al2O3 layer could be freely tuned from 50 μm to 300 μm (Fig. S1, ESI†). For the alumina-coated egg-shell Pd/α-Al2O3@SiC catalyst, the thickness of the Al2O3 layer was determined by the amount of Al2O3 loading on the SiC substrate.
As seen in Fig. 7, we explored the catalytic performances of the Pd/α-Al2O3@SiC catalysts with different Al2O3 loadings in acetylene selective hydrogenation. The performances of the Pd/α-Al2O3@SiC catalysts for the selective hydrogenation of acetylene to ethylene were generally excellent overall. The acetylene conversion increased as the Al2O3 loading on SiC increased, however, the tendency was not significant when the Al2O3 loading on SiC was more than 9.0 wt%. This means that Pd active sites are well dispersed on the Al2O3 layer when the thickness of the Al2O3 layer on the SiC substrate is more than 110 ± 10 μm. Moreover, the ethylene selectivity gradually decreased as the acetylene conversion increased, consistent with previous reports for acetylene selective hydrogenation catalyzed by supported Pd catalysts.35 This is because the reaction of acetylene hydrogenation is a typical consecutive reaction and ethylene is produced as an intermediate.
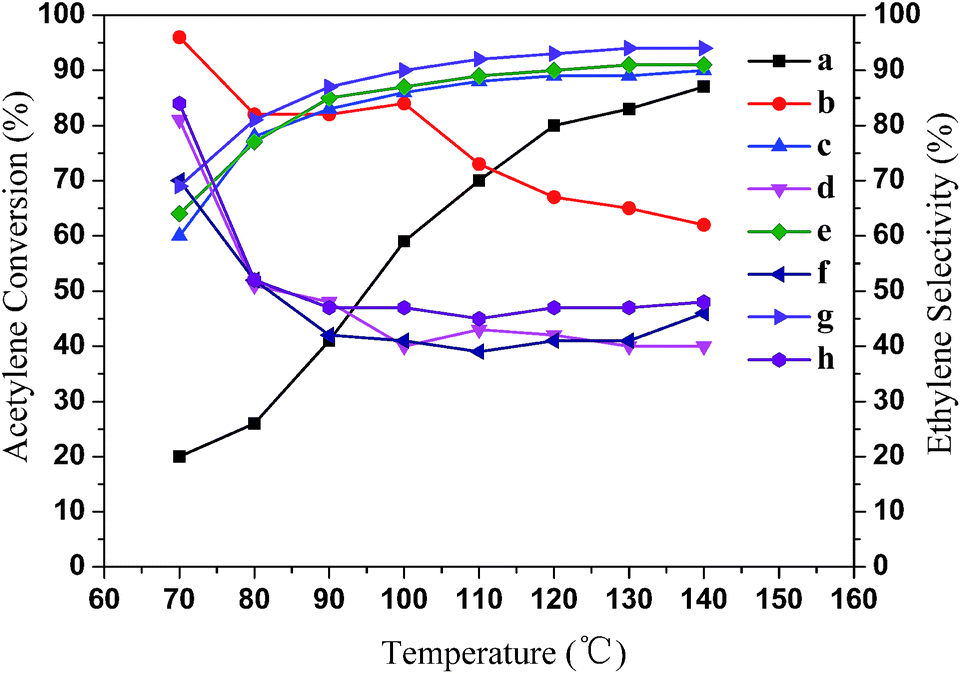 | ||
| Fig. 7 Performances of the egg-shell Pd/α-Al2O3@SiC catalysts at atmospheric pressure. Acetylene conversion: (a) 7.3 wt% Al2O3 loading, (c) 9.0 wt% Al2O3 loading, (e) 12.0 wt% Al2O3 loading and (g) 14.1 wt% Al2O3 loading. Ethylene selectivity: (b) 7.3 wt% Al2O3 loading, (d) 9.0 wt% Al2O3 loading, (f) 12.0 wt% Al2O3 loading and (h) 14.1 wt% Al2O3 loading. (H2/C2H2 = 1.6, 0.71% H2, 0.45% C2H2, 7.15% C2H6, and balanced C2H4.) | ||
Generally, for supported Pd catalysts, Ag has been used to modify the catalyst as an excellent promoter for achieving high ethylene selectivity.25–27 According to the results of an adhesion test and an abrasion resistance test, the Al2O3 layer of a Pd–Ag/α-Al2O3@SiC catalyst (14.1 wt% Al2O3 loading on SiC) has good adhesion and abrasion resistance (Tables S1 and S2, ESI†). Therefore, we chose the 14.1 wt% Al2O3 loading on the SiC substrate to prepare a Pd–Ag/α-Al2O3@SiC catalyst for enhancing ethylene selectivity. The results of a selectivity comparison of different Pd catalysts based on similar conversion levels are shown in Table 2. Industrially, in the tail-end hydrogenation process, acetylene must be completely removed from ethylene-rich streams. Therefore, a slight excess of hydrogen is added to the gas feed streams and the quantity of the H2/C2H2 (mole ratio) is larger than one. The selectivity of the catalyst beds is mainly controlled by the value of the H2/C2H2 ratio and the temperature.12 In general, ethylene selectivity will decrease as the reaction temperature increases at the same value of H2/C2H2. However, the α-Al2O3@SiC composite material has a better thermal stability than a conventional pellet α-Al2O3 support. Entries 1–4 in Table 2 show a comparison between the α-Al2O3@SiC composite material and a conventional pellet α-Al2O3 support. For identical Pd loading, the ethylene selectivities of the Pd/α-Al2O3@SiC catalyst and the Pd–Ag/α-Al2O3@SiC catalyst at 65 °C were respectively higher than those of a Pd/α-Al2O3 catalyst and a Pd–Ag/α-Al2O3 catalyst at 55 °C when the value of H2/C2H2 was 1.6. The α-Al2O3@SiC support was effective in enhancing ethylene selectivity when the acetylene conversions were at similar levels. For different Pd loadings, the literature describes a Pd catalyst supported on Zn-modified α-Al2O3 for the selective hydrogenation of acetylene in ethylene feed stock which was investigated by Chinayon et al.36 The value of H2/C2H2 was 1.1 and the Pd loading was 0.3 wt%. Its ethylene selectivity was 64% when the acetylene conversion was 82% (Table 2, entry 7). In contrast, for the Pd/α-Al2O3@SiC catalyst, when the value of H2/C2H2 was 1.6 and the Pd loading was 0.035 wt%, the ethylene selectivity was 65% and the acetylene conversion was 83% (Table 2, entry 6). For another example, the catalytic performance of a Pd–Ga/MgO–Al2O3 catalyst was reported by He et al.22 The value of H2/C2H2 was 2.0 and the Pd loading was 0.05 wt%. The acetylene conversion and ethylene selectivity were respectively 99% and 54% at 55 °C (Table 2, entry 5). For the Pd–Ag/α-Al2O3@SiC catalyst, the value of H2/C2H2 was 1.6 and the Pd loading was 0.035 wt%. Its ethylene selectivity was 57% at 65 °C when the acetylene conversion was 99% (Table 2, entry 3). Thus the ethylene selectivity of the Pd–Ag/α-Al2O3@SiC catalyst was a little higher than that of the Pd–Ga/MgO–Al2O3 catalyst.
| Entry | T/°C | Catalyst | Pd (wt%) | Conversion (%) | Selectivity (%) |
|---|---|---|---|---|---|
| a Egg-shell Pd catalyst with 14.1 wt% Al2O3 loading on SiC, H2/C2H2 = 1.6, 0.71% H2, 0.45% C2H2, 7.15% C2H6, and balanced C2H4, reaction pressure of 2 MPa.b Pd catalyst prepared with conventional extrusive pellet α-Al2O3 support, H2/C2H2 = 1.6, 0.71% H2, 0.45% C2H2, 7.15% C2H6, and balanced C2H4, reaction pressure of 2 MPa.c Egg-shell Pd catalyst with 7.3 wt% Al2O3 loading on SiC, H2/C2H2 = 1.6, 0.71% H2, 0.45% C2H2, 7.15% C2H6, and balanced C2H4, atmospheric pressure.d Literature data.22e Literature data.36 | |||||
| 1a | 65 | Pd/α-Al2O3@SiC | 0.035 | 95 | 48 |
| 2b | 55 | Pd/α-Al2O3 | 0.035 | 99 | 36 |
| 3a | 65 | Pd–Ag/α-Al2O3@SiC | 0.035 | 99 | 57 |
| 4b | 55 | Pd–Ag/α-Al2O3 | 0.035 | 100 | 41 |
| 5d | 55 | Pd–Ga/MgO–Al2O3 | 0.05 | 99 | 54 |
| 6c | 130 | Pd/α-Al2O3@SiC | 0.035 | 83 | 65 |
| 7e | 100 | Pd/Zn-modified-α-Al2O3 | 0.3 | 82 | 64 |
For assessing the initial performance, a side-line experiment for the selective hydrogenation of acetylene to ethylene was performed to contrast the Pd–Ag/α-Al2O3@SiC catalyst and the conventional pellet Pd–Ag/α-Al2O3 catalyst under industrial reaction conditions. A condition of this experiment is to thoroughly remove acetylene in an ethylene-rich stream and contrast ethylene selectivity with time on stream. In order to achieve this objective, the inlet temperature of the reactor for the Pd–Ag/α-Al2O3@SiC catalyst was kept at 65 °C and the inlet temperature of the reactor for the conventional pellet Pd–Ag/α-Al2O3 catalyst was kept at 55 °C. As shown in Fig. 8, the acetylene conversion of the conventional pellet Pd–Ag/α-Al2O3 catalyst was kept at 100% within the first 60 h which showed the stable initial activity of the catalyst. Its selectivity to ethylene was around 41%. The performance of the Pd–Ag/α-Al2O3@SiC catalyst was also stable within the first 60 h at 65 °C. Its acetylene conversion was kept at almost 99% and its selectivity to ethylene was around 57%, which was 16% more than that of the conventional pellet Pd–Ag/α-Al2O3 catalyst (Fig. 8). As can be seen in Fig. 8, this indicates that the Pd–Ag/α-Al2O3@SiC catalyst had a good initial performance without any noticeable deactivation within the first 60 h. The fluctuation of ethylene selectivity can be observed due to the dynamic change of the value of H2/C2H2 between 1.5 and 1.6. In our present work, although the performance of the Pd–Ag/α-Al2O3@SiC catalyst still needs to be improved further, it is a promising candidate for the selective hydrogenation of acetylene to ethylene.
 | ||
| Fig. 8 Side-line experiment results, in acetylene conversion and ethylene selectivity data show percentage of error bar: (a) acetylene conversion of Pd–Ag/Al2O3 catalyst at 55 °C, (b) ethylene selectivity of Pd–Ag/α-Al2O3 catalyst at 55 °C, (c) acetylene conversion of Pd–Ag/α-Al2O3@SiC catalyst at 65 °C and (d) ethylene selectivity of Pd–Ag/α-Al2O3@SiC catalyst at 65 °C. (14.1 wt% Al2O3 loading on SiC, H2/C2H2 = 1.6, 0.71% H2, 0.45% C2H2, 7.15% C2H6, and balanced C2H4, reaction pressure of 2 MPa.) | ||
4. Conclusions
An alumina-coated, egg-shell Pd/α-Al2O3@SiC catalyst – comprising a thickness-controlled Al2O3 layer on SiC – was studied by XRD, NH3-TPD, mercury porosimetry, SEM and TEM. In summary, we demonstrated a facile approach for preparing the egg-shell α-Al2O3@SiC composite material using a one-step spray coating method. The egg-shell α-Al2O3@SiC composite material was utilized as a support for the selective hydrogenation of acetylene to ethylene. The thickness of the α-Al2O3 layer could be freely tuned from 50 μm to 300 μm. The newly designed α-Al2O3@SiC support could very effectively control the Pd precursor penetration range, to achieve uniform distribution of Pd active sites on the Al2O3 layer. As compared with a conventional pellet Pd–Ag/α-Al2O3 catalyst, the Pd–Ag/α-Al2O3@SiC catalyst had a higher ethylene selectivity based on the same acetylene conversion, and is therefore a promising candidate for the selective hydrogenation of acetylene to ethylene.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51404097), the Sinopec Beijing Research Institute of Chemical Industry (11-08ZS0442), the Program for Innovative Research Team (in Science and Technology) in the University of Henan Province (16IRTSTHN005) and the Doctoral Foundation of Henan Polytechnic University (B2015-15 and B2015-12).Notes and references
- I. L. C. Buurmans and B. M. Weckhuysen, Nat. Chem., 2012, 4, 873–886 CrossRef CAS PubMed.
- C. Perego and R. Millini, Chem. Soc. Rev., 2013, 42, 3956–3976 RSC.
- C. M. A. Parlett, K. Wilson and A. F. Lee, Chem. Soc. Rev., 2013, 42, 3876–3893 RSC.
- T. Rodenas, I. Luz, G. Prieto, B. Seoane, H. Miro, A. Corma, F. Kapteijn, F. X. Llabrés i Xamena and J. Gascon, Nat. Mater., 2015, 14, 48–55 CrossRef CAS PubMed.
- W. Yang, X. Liu, X. Yue, J. Jia and S. Guo, J. Am. Chem. Soc., 2015, 137, 1436–1439 CrossRef CAS PubMed.
- Y. Wang, Y. Lai, L. Song, Z. Zhou, J. Liu, Q. Wang, X. Yang, C. Chen, W. Shi, Y. Zheng, M. Rauf and S. Sun, Angew. Chem., Int. Ed., 2015, 54, 9907–9910 CrossRef CAS PubMed.
- X. Zhou, J. Qiao, L. Yang and J. Zhang, Adv. Energy Mater., 2014, 4, 1301523, DOI:10.1002/aenm.201301523.
- X. Kong, C. Chen and Q. Chen, Chem. Soc. Rev., 2014, 43, 2841–2857 RSC.
- H. Shi, Y. Shen, F. He, Y. Li, A. Liu, S. Liu and Y. Zhang, J. Mater. Chem. A, 2014, 2, 15704–15716 CAS.
- W. Kiciński, M. Szala and M. Bystrzejewski, Carbon, 2014, 68, 1–32 CrossRef.
- A. Molnár, A. Sárkány and M. Varga, J. Mol. Catal. A: Chem., 2001, 173, 185–221 CrossRef.
- A. Borodziński and G. C. Bond, Catal. Rev.: Sci. Eng., 2006, 48, 91–144 Search PubMed.
- A. Borodziński and G. C. Bond, Catal. Rev.: Sci. Eng., 2008, 50, 379–469 Search PubMed.
- I. Y. Ahn, J. H. Lee, S. S. Kum and S. H. Moon, Catal. Today, 2007, 123, 151–157 CrossRef CAS.
- I. Y. Ahn, J. H. Lee, S. S. Kum and S. H. Moon, Appl. Catal., A, 2009, 360, 38–42 CrossRef CAS.
- B. Yang, R. Burch, C. Hardacre, G. Headdock and P. Hua, J. Catal., 2013, 305, 264–276 CrossRef CAS.
- S. Riyapan, Y. Boonyongmaneerat, O. Mekasuwandumrong, P. Praserthdam and J. Panpranot, Catal. Today, 2015, 245, 134–138 CrossRef CAS.
- B. Zhu and B. W.-L. Jang, J. Mol. Catal. A: Chem., 2014, 395, 137–144 CrossRef CAS.
- R. Hou, X. Lan and T. Wang, Catal. Today, 2015, 251, 47–52 CrossRef CAS.
- A. J. McCue, C. J. McRitchie, A. M. Shepherd and J. A. Anderson, J. Catal., 2014, 319, 127–135 CrossRef CAS.
- S. K. Kim, C. Kim, J. H. Lee, J. Kim, H. Lee and S. H. Moon, J. Catal., 2013, 306, 146–154 CrossRef CAS.
- Y. He, L. Liang, Y. Liu, J. Feng, C. Ma and D. Li, J. Catal., 2014, 309, 166–173 CrossRef CAS.
- A. D. Benavidez, P. D. Burton, J. L. Nogales, A. R. Jenkins, S. A. Ivanov, J. T. Miller, A. M. Karim and A. K. Datye, Appl. Catal., A, 2014, 482, 108–115 CrossRef CAS.
- Y. Zhang, W. Diao, C. T. Williams and J. R. Monnier, Appl. Catal., A, 2014, 469, 419–426 CrossRef CAS.
- A. Pachulski, R. Schödel and P. Claus, Appl. Catal., A, 2011, 400, 14–24 CrossRef CAS.
- W. Huang, J. R. McCormick, R. F. Lobo and J. G. Chen, J. Catal., 2007, 246, 40–51 CrossRef CAS.
- C. Shi, R. Hoisington and B. W. L. Jang, Ind. Eng. Chem. Res., 2007, 46, 4390–4395 CrossRef CAS.
- J. Panpranot, K. Kontapakdee and P. Praserthdam, Appl. Catal., A, 2006, 314, 128–133 CrossRef CAS.
- O. Mekasuwandumrong, N. Wongwaranon, J. Panpranot and P. Praserthdam, Mater. Chem. Phys., 2008, 111, 431–437 CrossRef CAS.
- E. Esmaeili, A. M. Rashidi, A. A. Khodadadi, Y. Mortazavi and M. Rashidzadeh, Fuel Process. Technol., 2014, 120, 113–122 CrossRef CAS.
- A. Sárkány, A. Horváth and A. Beck, Appl. Catal., A, 2002, 229, 117–125 CrossRef.
- S. K. Kim, J. H. Lee, I. Y. Ahn, W. J. Kim and S. H. Moon, Appl. Catal., A, 2011, 401, 12–19 CrossRef CAS.
- W. J. Kim, J. H. Kang, I. Y. Ahn and S. H. Moon, Appl. Catal., A, 2004, 268, 77–82 CrossRef CAS.
- M. Armbrüster, K. Kovnir, M. Behrens, D. Teschner, Y. Grin and R. Schlögl, J. Am. Chem. Soc., 2010, 132, 14745–14747 CrossRef PubMed.
- J. H. Kang, E. W. Shin, W. J. Kim, J. D. Park and S. H. Moon, J. Catal., 2002, 208, 310–320 CrossRef CAS.
- S. Chinayon, O. Mekasuwandumrong, P. Praserthdam and J. Panpranot, Catal. Commun., 2008, 9, 2297–2302 CrossRef CAS.
- W. J. Kim, I. Y. Ahn, J. H. Lee and S. H. Moon, Catal. Commun., 2012, 24, 52–55 CrossRef CAS.
- I. Y. Ahn, W. J. Kim and S. H. Moon, Appl. Catal., A, 2006, 308, 75–81 CrossRef CAS.
- M. M. Johnson, D. W. Walker and G. P. Nowack, US Pat., 4404124, 1983.
- S. H. Brown and T. P. Cheung, US Pat., 6127310, 2000.
- S. Xie, X. Tong, G. Jin, Y. Qin and X. Guo, J. Mater. Chem. A, 2013, 1, 2104–2109 CAS.
- Y. Zhang, J. Zang, L. Dong, X. Cheng, Y. Zhao and Y. Wang, J. Mater. Chem. A, 2014, 2, 10146–10153 CAS.
- Z. Chen, F. Bing, Q. Liu, Z. Zhang and X. Fang, J. Mater. Chem. A, 2015, 3, 4652–4658 CAS.
- Y. Peng, Z. Guo, J. Yang, D. Wang and W. Yuan, J. Mater. Chem. A, 2014, 2, 6296–6300 CAS.
- X. Guo, X. Tong, Y. Wang, C. Chen, G. Jin and X. Guo, J. Mater. Chem. A, 2013, 1, 4657–4661 CAS.
- Y. Li, Z. Yu, J. Meng and Y. Li, Int. J. Hydrogen Energy, 2013, 38, 3898–3904 CrossRef CAS.
- A. R. de la Osa, A. De Lucas, J. Díaz-Maroto, A. Romero, J. L. Valverde and P. Sánchez, Catal. Today, 2012, 187, 173–182 CrossRef CAS.
- X. Zhu, X. Lu, X. Liu, D. Hildebrandt and D. Glasser, Chem. Eng. J., 2014, 247, 75–84 CrossRef CAS.
- J. M. García-Vargas, J. L. Valverde, J. Díez, P. Sánchez and F. Dorado, Appl. Catal., B, 2015, 164, 316–323 CrossRef.
- R. Dhiman, S. N. Stamatin, S. M. Andersen, P. Morgen and E. M. Skou, J. Mater. Chem. A, 2013, 1, 15509–15516 CAS.
- R. Dhiman, E. Johnson, E. M. Skou, P. Morgen and S. M. Andersen, J. Mater. Chem. A, 2013, 1, 6030–6036 CAS.
- Y. Liu, I. Florea, O. Ersen, C. Pham-Huu and C. Meny, Chem. Commun., 2015, 51, 145–148 RSC.
- W. Song, Z. Chen, C. Yang, Z. Yang, J. Tai, Y. Nan and H. Lu, J. Mater. Chem. A, 2015, 3, 1049–1057 CAS.
- B. Liu, J. Du, X. Lv, Y. Qiu and C. Tao, Catal. Sci. Technol., 2015, 5, 1241–1250 CAS.
- F. Ding, A. Zhang, M. Liu, X. Guo and C. Song, RSC Adv., 2014, 4, 8930–8938 RSC.
- Y. Deng, W. Zhou, H. Lv, Y. Zhang, C. Au and S. Yin, RSC Adv., 2014, 4, 37296–37301 RSC.
- X. Qian, D. Xiong, A. M. Asiri, S. B. Khan, M. M. Rahman, H. Xu and D. Zhao, J. Mater. Chem. A, 2013, 1, 7525–7532 CAS.
- W. Qin, K. Guan, B. Lei, Y. Liu, C. Peng and J. Wu, J. Membr. Sci., 2015, 490, 160–168 CrossRef CAS.
- W. Qin, B. Lei, C. Peng and J. Wu, Mater. Lett., 2015, 144, 74–77 CrossRef CAS.
- N. Wongwaranon, O. Mekasuwandumrong, P. Praserthdam and J. Panpranot, Catal. Today, 2008, 131, 553–558 CrossRef CAS.
- B. M. Nagaraja, H. Jung, D. R. Yang and K.-D. Jung, Catal. Today, 2014, 232, 40–52 CrossRef CAS.
- S. Komhom, O. Mekasuwandumrong, P. Praserthdam and J. Panpranot, Catal. Commun., 2008, 10, 86–91 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM photographs of the Al2O3 layer at the different Al2O3 loadings on SiC, detailed analysis of the adhesion test and abrasion resistance test. See DOI: 10.1039/c6ra08320j |
| This journal is © The Royal Society of Chemistry 2016 |