DOI:
10.1039/C6RA10832F
(Paper)
RSC Adv., 2016,
6, 57163-57173
Direct C–H arylation for various Ar-cored diketopyrrolopyrrole containing small molecules in solution-processed field-effect transistors†
Received
27th April 2016
, Accepted 7th June 2016
First published on 8th June 2016
Abstract
Direct (hetero) C–H arylation is an advantageous tool for the synthesis of diketopyrrolopyrrole (DPP) derivatives, because of fewer synthetic steps, better atom economy and being environmentally friendly. Herein, four diketopyrrolopyrrole containing linear structured D–A–π–A–D small molecules Ar(DPPT2)2 are facilely synthesized in high yields of 73–82% through C–H direct arylation, where Ar (from electron-donating 1,4-phenyl, 1,4-naphthyl or 9,10-anthryl to electron-accepting 2,5-pyridyl) is functionalized as the core π-bridge structure, DPP as the arm and bithiophene as end-groups. The dihedral angles between the central aryl rings and DPPT2 arms for the optimized geometries of Ph(DPPT2)2, NA(DPPT2)2 and AN(DPPT2)2 gradually increases from 20.3, 44.7 to 93.1°, respectively, which is 18.7° for the pyridyl cored Py(DPPT2)2. It is found that the optoelectronic properties can be elaborately tuned by variation of the central aryl bridge. Moreover, the coplanarity of the molecules as well as the electronic properties of central Ar units significantly affect the charge transport properties. Ph(DPPT2)2 possessing the best conjugated backbone planarity exhibits the highest hole mobility of 0.12 cm2 V−1 s−1 among the three Ar(DPPT2)2 compounds based on the electron-donating phenyl, naphthyl and anthryl cores, while the electron-withdrawing pyridyl core Py(DPPT2)2 shows a poor hole mobility of 6.47 × 10−4 cm2 V−1 s−1 despite of it having the most planar structure.
Introduction
In recent years, the industrial pigment, diketopyrrolopyrrole (DPP) has received considerable attention for constructing high performance materials for both organic field-effect transistors (OFETs) and organic photovoltaics (OPVs).1–7 DPP exhibits a coplanar geometry and can form hydrogen bonds with the neighboring units in the backbone, which favors intermolecular π–π stacking to facilitate the charge transport performance. Through judicious molecular design, DPP has emerged as one of the most promising building blocks for efficient solution-processed OFETs materials.8 Specifically, DPP containing conjugated polymers exhibited excellent OFET performance with a hole mobility above 13 cm2 V−1 s−1 and an electron mobility of 7 cm2 V−1 s−1.3,9–13 However, the charge transport properties of DPP based small molecules have been less studied, even though small molecular materials exhibit distinct advantages, such as more straight forward synthesis, versatile chemical modifications, well-defined structures as well as less batch-to-batch variations.14–16 Furthermore, the specific structure and confirmed molecular weight of small molecules enables better understanding of structure–property relationships and thus provides guidance to the design of high-performance semiconducting materials. Most DPP containing small molecules exhibit p-type behavior with low hole mobilities in the orders of 10−5 to 10−3 cm2 V−1 s−1.17–27 And few examples of solution-processed DPP-containing small molecules with hole mobility above 0.1 cm2 V−1 s−1 have been reported.21–23
In previous studies, most of DPP derivatives were commonly synthesized by Stille, Suzuki or Negishi cross-coupling reactions,24–49 where one arene is substituted with a functional group (Br, I, OTf, etc.), and the other contains an organometallic moiety such as –SnR3 and –ZnR or –B(OR)3, etc. However, integration of these functional groups entails extra synthetic steps and the compounds are normally unstable and/or toxic.50 Alternatively, DPP containing thiophene derivatives are ideal candidates for the exploration of direct arylation reactions, due to the ease of palladation through a concerted metalation–deprotonation (CMD) pathway.51–54 Therefore, the DPP derivatives are then easily accessible for Pd-catalyzed C–H arylation. Despite the significant progress in the synthesis of DPP derivatives, only limited examples have been reported on direct C–H activation.55–57
Herein, we report the design and synthesis of four DPP containing D–A–π–A–D small molecules Ar(DPPT2)2 by using various aromatics such as electron-donating 1,4-phenyl, 1,4-naphthyl or 9,10-anthryl or electron-accepting 2,5-pyridyl as the central Ar π-conjugated bridge, the electron-accepting (A) DPP as arm and the electron-donating (D) bithiophene as end-groups. All compounds could be facilely synthesized in good yields through the C–H direct arylation. Ar(DPPT2)2 with different central Ar π-bridges particularly with fused benzene or biaxially extended benzene moieties may offer opportunities for better understanding the correlation between the molecular geometry, optoelectronic properties, film morphology, and field-effect mobilities of DPP-based D–A–π–A–D small molecules. By introducing bithiophene end-group to the electron-withdrawing DPP block, the conjugation length of molecular backbone can be increased, resulting in the enhancement of intermolecular π–π interactions and the improvement of molecular packing as well as the charge carrier mobilities.58,59 The morphology of thin films of these materials was investigated by atomic force microscopy (AFM) and the film structures was analyzed by X-ray diffraction (XRD) and grazing incidence wide-angle X-ray scattering (GIWAXS). Their charge-carrier mobilities were extracted from the electrical characteristics of the field-effect transistors. By combining the experimental results of all these characterizations, we achieved a better understanding of structure–property relationships. Furthermore, the experimental results reveal that the Ph(DPPT2)2 is a promising candidate for high-performance solution-processed OFETs.
Experimental section
Materials
All reagents were purchased from commercial sources (Energy Chemical, Suna Tech Inc, Stream and Sigma Aldrich) and used without further purification. Reagent grade solvents used in this study were freshly dried using standard distillation methods.
Measurements
1H NMR and 13C NMR spectra were measured on a Bruker DRX-400 spectrometer. Elemental analysis (C, H and N) was performed using a Vario EL III microanalyzer. MALDI-TOF mass spectra were measured on a Bruker autoflex matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) spectrometer. UV-vis spectra were recorded on a Shimadzu UV-2500 recording spectrophotometer. Thermogravimetric analysis (TGA) was undertaken with a NETZSCH STA 449C instrument, and measurements were performed within the temperature ranged from 25 to 600 °C, heating at a rate of 20 °C min−1 under N2 atmosphere. Differential scanning calorimetry (DSC) was performed on a NETZSCH DSC 200 PC unit within the temperature range of −50 to 300 °C, heating at a rate of 10 °C min−1 under N2 atmosphere. Cyclic voltammetry (CV) was measured on a CHI660D electrochemical workstation using a solution of tetrabutylammonium hexafluorophosphate (Bu4NPF6) in dichloromethane (0.1 mol L−1) as the electrolyte, a platinum plate as the working electrode, a platinum wire as the auxiliary electrode, and a silver wire as the pseudo-reference electrode, with ferrocenium-ferrocene (Fc+/Fc) as the internal standard. Atomic force microscopy (AFM) was conducted on SPA 300HV machine in tapping mode using an SPI3800 controller, Seiko Instruments Industry, Co., Ltd. X-ray diffraction measurements were performed using a D8 Bruker setup (λ = 1.5406 Å) and a Lynx Eye linear detector in the q-range 0.3–1.3 Å−1. Grazing incidence wide angle X-ray scattering (GIWAXS) measurements were performed at beamline I07, Diamond Light Source, Harwell Campus, Didcot UK, using a Pilatus 2 M detector (λ = 0.992 Å) under a protective atmosphere to avoid beam damage.
OFET device fabrication
Devices were fabricated in a top-gate, bottom-contact (TG-BC) configuration on glass substrates (Corning 7059). Chromium/gold (3 nm/12 nm, respectively) source and drain electrodes were defined by photolithography. Thin films of the active layer were spin cast at 2000 rpm from o-dichlorobenzene (o-DCB) solutions (ca. 5 mg mL−1) and annealed at 120 °C for 10 min in a glovebox. A poly(perfluoroalkenylvinyl ether) (Cytop) or poly(methyl methacrylate) (PMMA) dielectric were spin-coated onto the semiconductor in glovebox under nitrogen atmosphere and then baked at 90 °C for 20 minutes before gold were evaporated through a shadow mask as gate (channel length L = 20 μm and width W = 1 mm). The thickness of PMMA and Cytop layers are about 480 nm and 550 nm, respectively. And Cytop was dissolved in a fluorinated solvent and PMMA was dissolved in n-butyl acetate. The corresponding capacitance values were calculated by using Ci = εε0S/d. As PMMA solution can dissolve the two active materials of NA(DPPT2)2 and Py(DPPT2)2, devices with PMMA as dielectrics were only fabricated for Ph(DPPT2)2. The OFET device characteristics were measured using an Agilent 4155B semiconductor parameter analyzer. The mobility was determined in the saturation regime using the following equation: IDS = (μWCi/2L)(VG − VT)2 where IDS is the drain–source current, μ is the field-effect mobility, W is the channel width, L is the channel length, Ci is the capacitance per unit area of the gate dielectric layer and VT is the threshold voltage.
Synthesis
The general synthetic route towards Ph(DPPT2)2, NA(DPPT2)2, AN(DPPT2)2 and Py(DPPT2)2 is outlined in Scheme 1 with detailed synthetic procedures given by Ph(DPPT2)2.
 |
| | Scheme 1 Synthetic route towards Ar(DPPT2)2. | |
6,6′-(5,5′-(1,4-Phenylene)bis(thiophene-5,2-diyl))bis(2,5-bis(2-ethylhexyl)-3-(5′′-hexyl-[2,2′:5′,2′′-terthiophen]-5-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione) (Ph(DPPT2)2). 2,5-Bis(2-ethylhexyl)-3-(500-hexyl-[2,20:50,200-terthiophen]-5-yl)-6-(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione (DPPT2) (164 mg, 0.22 mmol), 1,4-dibromobenzene (25.5 mg, 0.11 mmol), anhydrous K2CO3 (373 mg, 0.27 mmol), PivOH (29.2 mg, 0.032 mmol) and Pd(OAc)2 (4.5 mg, 0.022 mmol) were stirred in anhydrous dimethylacetamide (DMA, 5 mL) at 110 °C for 14 h under a nitrogen atmosphere. After cooling to room temperature, the mixture was poured into an aqueous solution of NaCl (200 mL) to remove any inorganic salts and the high boiling point solvent of DMA. The precipitate was extracted with CH2Cl2, washed with brine, dried over anhydrous MgSO4, and concentrated under reduced pressure. The crude product was purified by chromatography on silica gel (eluent: dichloromethane) and washed with MeOH to yield the title compound as a black solid (140 mg, 80%). 1H NMR (400 MHz, CDCl3): δ (ppm) 9.00 (d, J = 4.0 Hz, 2H), 8.96 (d, J = 4.0 Hz, 2H), 7.51 (m, 4H), 7.39 (d, J = 4 Hz, 2H), 7.16 (d, J = 4 Hz, 2H), 7.07 (d, J = 3.6 Hz, 2H), 6.95 (m, 4H), 6.64 (d, J = 3.6 Hz, 2H), 4.02 (m, 8H), 2.79 (m, 4H), 2.20 (m, 4H), 1.38 (m, 42H), 0.95 (m, 36H). N.B. We are unable to report the 13C NMR spectra of Ph(DPPT2)2 due to its tendency to aggregate in CDCl3 solution resulting in broad, unresolved signals. MS (MALDI-TOF) calcd for C94H114N4O4S8: 1619.66. Found: 1618.26 (M + H+). Anal. calcd for C94H114N4O4S8: C, 69.67; H, 7.09; N, 3.46. Found: C, 69.94; H, 7.09; N, 3.41.
6,6′-(5,5′-(Naphthalene-1,4-diyl)bis(thiophene-5,2-diyl))bis(2,5-bis(2-ethylhexyl)-3-(5′′-hexyl-[2,2′:5′,2′′-terthiophen]-5-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione) (NA(DPPT2)2). NA(DPPT2) was synthesized following the same procedure as Ph(DPPT2)2 by using 1,4-dibromonaphthalene (30.3 mg, 0.11 mmol) as the starting material to yield the title compound as a black solid (133 mg, 75%). 1H NMR (400 MHz, CDCl3): δ (ppm) 9.06 (d, J = 4.0 Hz, 2H), 8.99 (d, J = 4.0 Hz, 2H), 8.39 (d, J = 9.6 Hz, 2H), 7.63 (s, 2H), 7.48 (d, J = 4 Hz, 2H), 7.30 (d, J = 4.4 Hz, 2H), 7.25 (s, 2H), 7.22 (d, J = 3.6 Hz, 2H), 7.05 (m, 4H), 6.71 (d, J = 3.6 Hz, 2H), 4.09 (m, 8H), 2.80 (t, J = 3.8 Hz, 4H), 1.95 (m, 4H), 1.33 (m, 42H), 0.90 (m, 36H). 13C NMR (100 MHz, CDCl3): δ (ppm) 161.8, 161.6, 149.8, 147.5, 142.9, 139.4, 139.1, 137.0, 135.9, 134.0, 132.3, 131.9, 130.3, 129.3, 127.9, 127.6, 126.3, 126.1, 125.9, 125.1, 124.5, 124.0, 123.9, 108.4, 108.2, 46.0, 39.3, 31.6, 31.5, 30.4, 30.3, 30.2, 29.7, 29.6, 29.3, 28.8, 28.6, 28.5, 27.2, 25.5, 23.7, 23.7, 23.2, 23.1, 22.7, 22.6, 14.1, 14.1, 10.6. MS (MALDI-TOF) calcd for C98H116N4O4S8: 1669.68. Found: 1669.59 (M+). Anal. calcd for C98H116N4O4S8: C, 70.46; H, 7.00; N, 3.35. Found: C, 70.34; H, 6.99; N, 3.35.
6,6′-(5,5′-(Anthracene-9,10-diyl)bis(thiophene-5,2-diyl))bis(2,5-bis(2-ethylhexyl)-3-(5′′-hexyl-[2,2′:5′,2′′-terthiophen]-5-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione) (AN(DPPT2)2). AN(DPPT2)2 was obtained as a black solid (133 mg, 73%) by using 9,10-dibromoanthracene (37.5 mg, 0.11 mmol) as the starting material. 1H NMR (400 MHz, CDCl3): δ (ppm) 9.20 (d, J = 4.0 Hz, 2H), 9.01 (d, J = 4.0 Hz, 2H), 7.96 (m, 4H), 7.49 (m, 4H), 7.42 (d, J = 3.6 Hz, 2H), 7.32 (d, J = 4.4 Hz, 2H), 7.24 (d, J = 3.6 Hz, 2H), 7.07 (m, 4H), 6.72 (d, J = 3.6 Hz, 2H), 4.10 (m, 8H), 2.80 (m, 4H), 1.95 (m, 4H), 1.33 (m, 42H), 0.90 (m, 36H). 13C NMR (100 MHz, CDCl3): δ (ppm) 162.0, 161.6, 146.5, 144.3, 142.9, 139.4, 139.1, 137.1, 135.8, 134.0, 131.6, 131.4, 131.2, 129.2, 127.9, 126.5, 126.4, 126.0, 126.1, 125.1, 124.5, 124.1, 123.9, 108.5, 108.3, 46.1, 39.3, 31.9, 31.6, 31.5, 30.5, 30.2, 29.8, 29.7, 29.6, 29.5, 29.3, 29.3, 28.8, 28.6, 25.5, 23.8, 23.5, 23.2, 23.0, 22.7, 22.6, 14.1, 14.1, 14.0, 10.5. MS (MALDI-TOF) calcd for C110H131N5O4S8: 1719.70. Found: 1720.63 (M + H+). Anal. calcd for C102H118N4O4S8: C, 71.20; H, 6.91; N, 3.26. Found: C, 71.02; H, 6.91; N, 3.16.
6,6′-(5,5′-(Pyridine-2,5-diyl)bis(thiophene-5,2-diyl))bis(2,5-bis(2-ethylhexyl)-3-(5′′-hexyl-[2,2′:5′,2′′-terthiophen]-5-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione) (Py(DPPT2)2). Py(DPPT2)2 was prepared by using 2,5-dibromopyridine (25.6 mg, 0.11 mmol) as reactant to afford the final compound as a black solid (144 mg, 82%). 1H NMR (400 MHz, CDCl3): δ (ppm) 9.01 (d, J = 4.0 Hz, 2H), 8.98 (d, J = 4.0 Hz, 2H), 7.76 (m, 3H), 7.61 (s, 1H), 7.59 (s, 1H), 7.27 (m, 2H), 7.19 (d, J = 4.0 Hz, 2H), 7.03 (m, 4H), 6.71 (d, J = 3.6 Hz, 2H), 4.12 (m, 8H), 2.80 (t, J = 3.8 Hz, 4H), 1.95 (m, 4H), 1.33 (m, 42H), 0.90 (m, 36H). 13C NMR (100 MHz, CDCl3): δ (ppm) 161.7, 161.5, 151.4, 148.7, 146.4, 142.9, 140.0, 139.3, 139.0, 137.5, 137.3, 136.5, 134.0, 131.6, 127.8, 126.0, 125.9, 125.0, 124.4, 124.0, 123.8, 108.9, 108.3, 46.1, 39.3, 39.3, 31.9, 31.6, 31.5, 30.4, 30.3, 30.2, 29.7, 29.6, 29.3, 28.8, 28.6, 28.5, 27.2, 23.7, 23.2, 23.1, 22.7, 22.6, 14.1, 14.1, 14.0, 10.7, 10.6. MS (MALDI-TOF) calcd for C93H113N5O4S8: 1620.66. Found: 1620.64 (M+). Anal. calcd for C93H113N5O4S8: C, 68.89; H, 7.02; N, 4.32. Found: C, 68.67; H, 7.04; N, 4.29.
Results and discussion
Synthesis and characterization
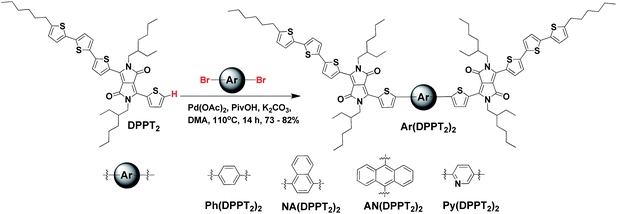
Scheme 1 shows the synthetic routes for the four DPP containing compounds. DPPT2 was synthesized and purified according to our previous work.60 All target small molecules were synthesized via a Pd-catalysed, direct C–H arylation reaction between DPPT2 and the corresponding dibrominated aromatic units. The resulting linear D–A–π–A–D small molecules, where DPP as arm (acceptor), bithiophene as end-groups (donor) and Ar (phenyl, naphthyl, anthryl and pyridyl) as the conjugated π-bridge or the core structure, were abbreviated as Ar(DPPT2)2. The reaction carried out under simple and ligandless conditions, gave the products Ar(DPPT2)2 in good yield of 72–83%. It is noted that the approach presented in this work directly linking aryl-H and aryl-Br reactants without cumbersome boronic acids or toxic tin reagents that are commonly used in other C–C bond forming reactions reduced the synthetic steps and environmental impact. The final products were soluble in common organic solvents, such as chloroform, chlorobenzene and o-dichlorobenzene at room temperature. And the chemical structure of the new compounds was confirmed by a combination of techniques of 1H NMR, 13C NMR, high-resolution mass spectroscopy and elemental analysis, which were in agreement with theoretical values.
Density functional theory (DFT) calculations were used to investigate the ground-state geometry and frontier molecular orbital electron density distributions of the four compounds. Calculations were performed using the Gaussian 09 suite at the B3LYP/6-311G level of theory.61 In order to simplify the calculations, alkyl-chain substituents were replaced with methyl groups. The optimized structures and frontier molecular orbital plots are illustrated in Fig. 1. As shown, DPPT2 exhibited almost planar structure, while the choice of the bridged aromatic unit was found to have a significant impact on the backbone planarity of the four compounds. In the case of Py(DPPT2)2 and Ph(DPPT2)2, a relatively planar backbone structure was predicted with small torsion angles of 20.3 and 18.7° between the bridging group and the flanking thiophene on the DPP unit, respectively. In contrast, the addition of the more sterically bulky naphthalene and anthracene core in NA(DPPT2)2 and AN(DPPT2)2 was predicted to lead to a highly twisted structure with large torsion angles of 44.7 and 93.1°, respectively. It is noted that the central anthracene core and the DPPT2 arm performed an orthogonal geometry which lead to the worst planarity in AN(DPPT2)2.
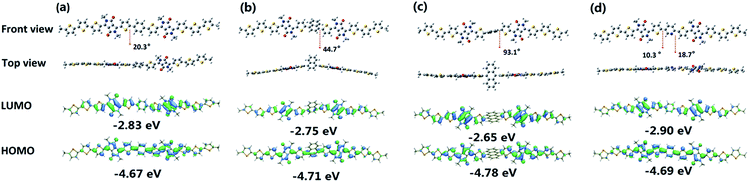 |
| | Fig. 1 Optimized geometries and frontier molecular orbital electron density distributions of (a) Ph(DPPT2)2, (b) NA(DPPT2)2, (c) AN(DPPT2)2 and (d) Py(DPPT2)2. Angles between adjacent ring planes are shown in front view. | |
As shown in the frontier molecular orbital electron density distributions, the highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbitals (LUMO) for Ph(DPPT2)2, NA(DPPT2)2 and Py(DPPT2)2 are delocalized almost over the entire molecular skeleton, which might potentially lead to strong π–π interactions and thus enhanced charge transport properties.62,63 Interestingly the HOMO and LUMO for AN(DPPT2)2 is only localized on the arm of DPPT2 units. It is found that all the compounds exhibited similar theoretical HOMO values (4.67–4.78 eV, Fig. 1), whereas, variations on LUMO levels were observed from 2.65–2.90 eV according to the electronic properties of the central aryl bridge. Py(DPPT2)2 with the electron-accepting pyridyl core which stabilizes and lowers the energy levels exhibited the lowest LUMO values.
Thermal properties
Thermogravimetric analysis (TGA) was used to evaluate the thermal stability of the compounds. As shown in Fig. 2, all four compounds exhibited good thermal stability, with decomposition temperatures (Td, 5% weight loss) occurring at 401, 386, 394 and 403 °C for Ph(DPPT2)2, NA(DPPT2)2, AN(DPPT2)2 and Py(DPPT2)2, respectively. Their thermal properties were also investigated by differential scanning calorimetry (DSC). As shown in Fig. 3, the four compounds exhibited an endothermic melt at 244, 169, 203 and 218 °C, respectively. Upon cooling, an significant exothermic crystallization peak was observed for Ph(DPPT2)2 at 217 °C (Tc). Interestingly, in the case of NA(DPPT2)2 and AN(DPPT2)2, an weak exothermic crystallization was observed at much lower temperatures of 170 and 147 °C, respectively. However, no obvious exothermic crystallization was detected in the DSC trace of the unsymmetrical Py(DPPT2)2. It is deduced that the lower decomposition temperatures and lower melting point (Tm) as well as weaker crystallization intensity for the more bulky naphthyl and anthryl containing NA(DPPT2)2 and AN(DPPT2)2, could be attributed to their more torsionally twisted structures, which reduced the intermolecular interaction compared to other compounds especially the phenyl containing Ph(DPPT2)2 due to their similar electronic properties in the central aryl core.
 |
| | Fig. 2 Thermal analysis of the four compounds. | |
 |
| | Fig. 3 DSC curves of (a) Ph(DPPT2)2, (b) NA(DPPT2)2, (c) AN(DPPT2)2 and (d) Py(DPPT2)2. | |
Optical and electrochemical properties
The optical properties of the four compounds were determined by UV-vis absorption spectroscopy. Normalized absorption spectra in dilute chloroform solution and solid thin film were shown in Fig. 4. The related key data was summarized in Table 1. In solution, all four compounds exhibited two distinct absorption bands between 300–483 nm and 483–723 nm, respectively. The shorter wavelength bands could be attributed to localized π–π* transitions on the donor or acceptor, while the longer wavelength bands could be originated from intramolecular charger transfer (ICT) transition between the electron donors and acceptors.64,65 The dihedral angles between thiophene rings in DPP and the central Ar cores increased progressively from Ph(DPPT2)2 to NA(DPPT2)2 and further to AN(DPPT2)2, which reduced the molecular π-conjugation. Therefore, the corresponding maximum absorption wavelength gradually blue shifted from 650 to 626 and further to 612 nm, respectively. Besides, a 13 and 9 nm blue-shift was observed for shoulder and peak absorption respectively, in Py(DPPT2)2 compared to Ph(DPPT2)2, due to different electronic properties of the central pyridine and benzene core. On the other hand, significantly red-shifted absorption was observed in the film state compared in solution. In addition, considerably broader longer wavelength absorption from 500–800 nm with obvious dual-band-absorbing property was achieved in the phenyl and pyridyl cored compounds due to their most planar structure for strong intermolecular interaction. And these two bands could be successively assigned to intramolecular charge transfer transition from electron donor to acceptor as well as intermolecular aggregation.58,66 Whereas, the anthryl cored AN(DPPT2)2 exhibited the sharpest absorption spectra because the perpendicular geometry impeded intermolecular aggregation.
 |
| | Fig. 4 Normalized UV-vis absorption spectra of the four compounds in (a) CHCl3 solution and (b) thin film. | |
Table 1 Thermal and optoelectronic properties of the four compounds
| Material |
Td (°C) |
Tm (°C) |
Tc (°C) |
λmax solna (nm) |
λmax filmb (nm) |
HOMOc (eV) |
LUMOc (eV) |
| Dilute chloroform solution. As-cast thin-films on glass substrates. Measured by CV. |
| Ph(DPPT2)2 |
401 |
244 |
217 |
610, 650 |
599, 662, 725 |
4.96 |
3.48 |
| NA(DPPT2)2 |
386 |
169 |
170 |
626 |
649, 695 |
4.97 |
3.26 |
| AN(DPPT2)2 |
394 |
203 |
147 |
612 |
656 |
5.01 |
3.24 |
| Py(DPPT2)2 |
403 |
218 |
— |
597, 641 |
618, 684 |
4.98 |
3.29 |
The electrochemical properties of the compounds in dichloromethane solution (0.2 mg mL−1) were investigated by cyclic voltammetry (CV). The cyclic voltammograms of the four compounds were shown in Fig. 5 and the data was summarized in Table 1. All four compounds exhibited reversible or quasi-reversible oxidation and reduction behavior. The HOMO and LUMO energy level were calculated using the following equations: EHOMO = −(EOX − EFc + 4.8) eV and ELUMO = −(ERED − EFc + 4.8) eV, where EOX and ERED were determined from the onset of the first potential of the oxidation and reduction curves, respectively, and EFc was taken as the half-wave potential of ferrocene. As shown in Table 1, we found that the nature of the bridging aromatic unit has little effect on the HOMO energy, which are all well below the limit for efficient hole injection from the gold electrodes (ca. 5.1 eV) in organic field effect devices.67
 |
| | Fig. 5 Cyclic voltammogram curves of (a) Ph(DPPT2)2, (b) NA(DPPT2)2, (c) AN(DPPT2)2 and (d) Py(DPPT2)2. | |
Thin film morphology
X-ray diffraction (XRD), grazing incidence wide angle X-ray scattering (GIWAXS) and atomic force microscopy (AFM) were used to investigate the microstructure and thin film morphology of the materials. Unfortunately, it is impossible to form a continuous thin film of AN(DPPT2)2 due to strong de-wetting of the solution on the glass substrate and as a result, we were unable to study the thin film morphology of this compound. The AFM topography images of spin-coated films of Ph(DPPT2)2, NA(DPPT2)2 and Py(DPPT2)2 were shown in Fig. 6. As shown, Ph(DPPT2)2 formed a relatively smooth and comparably amorphous thin film with a low root-mean-square (RMS) surface roughness of 1.1 nm. In contrast, both NA(DPPT2)2 and Py(DPPT2)2 exhibited significant rough surfaces with higher RMS values of 2.6 and 4.5 nm, respectively. In addition, a large amount of closely packed nano-ribbons with the size of about 50 nm could be found in Ph(DPPT2)2. However, the film morphology of NA(DPPT2)2 demonstrated more polycrystalline nature. When taking a closer observation at this film morphology, we could see there were fiber features in the films and these fibers exhibited quite good alignment in each clusters. The morphology of the Py(DPPT2)2 film (Fig. 6c) was rather different from the other two compounds which showed obvious grain structures typically for small molecule films.
 |
| | Fig. 6 AFM topography images (5 × 5 μm) of (a) Ph(DPPT2)2, (b) NA(DPPT2)2 and (c) Py(DPPT2)2 (the corresponding phase images are shown in Fig. S1†). | |
The thin film structure of the three materials was further investigated by X-ray diffraction. As shown in Fig. 7, all three compounds exhibited a first order, diffraction peak corresponding to an out-of-plane lattice with d-spacing of 1.44 nm for NA(DPPT2)2 and Py(DPPT2)2, and 1.49 nm for Ph(DPPT2)2, respectively. It is important to note that the intensity of this peak for Ph(DPPT2)2 was considerably lower than NA(DPPT2)2 and Py(DPPT2)2, and it is in accordance with the AFM images, in which Ph(DPPT2)2 showed most amorphous surfaces and the least domain sizes. The film structure of Ph(DPPT2)2 and NA(DPPT2)2 was also characterized by wide angle X-ray scattering (GIWAXS) (Fig. 8).
 |
| | Fig. 7 XRD results of Ph(DPPT2)2, NA(DPPT2)2 and Py(DPPT2)2. The out-of-plane lattice spacing can be extracted from the peaks. | |
 |
| | Fig. 8 GIWAXS reciprocal space maps of Ph(DPPT2)2 (top panel) and NA(DPPT2)2 (bottom panel) measured at an angle of incidence of 0.3°. | |
In the GIWAXS images of Ph(DPPT2)2 sample, only a Debye ring corresponding to the Q position reported for XRD and no clear in-plane Bragg-peak was detected. This observations indicate a weak in-plane long-range order and almost random orientation of the crystal grains (i.e. only weak texture), which is also consistent with the morphology discussed above. In contrast, the GIWAXS images of sample NA(DPPT2)2 showed a sharp in-plane Bragg-peak, corresponding to a π–π stacking of 0.39 nm. Multiple well defined Bragg-peaks displayed in GIWAXS of NA(DPPT2)2, indicated that the corresponding film was much more textured than that of Ph(DPPT2)2 and also demonstrated a significantly higher long-range order.
OFET performance
The charge transport behavior of the materials was investigated in top-gate, bottom-contact OFET devices source drain electrodes (Cr/Au = 3 nm/15 nm) were prepared by photolithography. In our initial investigations, a fluoropolymer dielectric, CYTOP was used. The respective transfer and output characteristics are shown in Fig. 9 and the data is summarized in Table 2. Unfortunately, we were unable to form a continuous thin film of AN(DPPT2)2 due to de-wetting of the solution on the glass substrate. Thus, it is impracticable to study the charge transport behavior of AN(DPPT2)2 in this work. Both Ph(DPPT2)2 and Py(DPPT2)2 demonstrated typical p-type transport with average hole mobilities of 0.085 and 6 × 10−4 cm2 V−1 s−1 respectively. Interestingly, slight ambipolar character in NA(DPPT2)2 (Fig. 9b) devices was observed which is in agreement with other similar materials.17,68 The hole mobility of Ph(DPPT2)2 and NA(DPPT2)2 with different central π-bridge was initially compared. It is found that Ph(DPPT2)2 system exhibited slightly higher hole mobility than 0.037 cm2 V−1 s−1 of NA(DPPT2)2 even though NA(DPPT2)2 film was observed to be more crystalline. This can be ascribed to several aspects. Firstly, due to the difference in molecular structure, Ph(DPPT2)2 demonstrates higher planarity of the conjugated backbone than NA(DPPT2)2. Therefore, the stronger intra-molecule charge transport is expected, which is supposed as the main reason for the higher hole mobility of Ph(DPPT2)2. Secondly, the lower surface roughness in Ph(DPPT2)2 film might contribute to the higher mobility values as surface roughness is critical to charge transport in top-gate devices since charge transport mainly occurs at the semiconductor–dielectric interface.69 Despite we failed to fabricate the films or even OFET devices of AN(DPPT2)2, it is still expected that the mobility of this compound would be further lower due to the above two reasons. It should be noted that Py(DPPT2)2 exhibited a comparable HOMO level and good crystalline film structure, and moreover, a comparatively flat molecular backbone that is similar to Ph(DPPT2)2, however, compared to Ph(DPPT2)2, a significantly lower hole mobility was achieved in the electron-withdrawing pyridyl bridged Py(DPPT2)2. Therefore, it can be concluded that the configuration of the molecule backbone is not the only factor determining the final device performance. The difference in the electronic properties of the central π-bridged aryl units for the two compounds also leads to the variation of charge carrier mobilities. By comparing the electrical mobility of Ph(DPPT2)2 and Py(DPPT2)2, we presented that the electron-donating phenyl ring is more suitable for hole transport than the electron-withdrawing pyridine unit.70 The above electronic and structural characterizations suggest that the π-bridge aryl units have pronounced influence on the molecular packing and film structures.
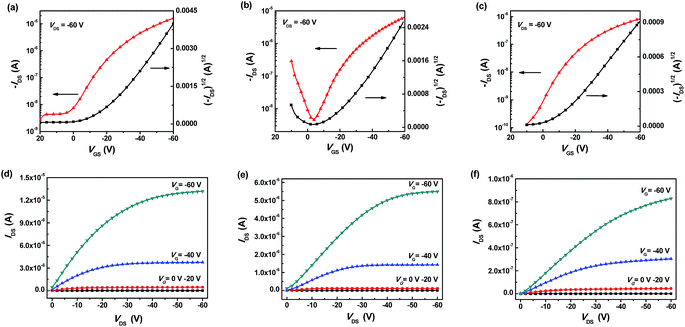 |
| | Fig. 9 Transfer (top) and output (bottom) plots (VDS = −60 V) of Ph(DPPT2)2 (a and d), NA(DPPT2)2 (b and e) and Py(DPPT2)2 (c and f) OFET devices annealed at 120 °C. | |
Table 2 OFET characteristics
| Material |
μavgc (cm2 V−1 s−1) |
μmax (cm2 V−1 s−1) |
Ion/Ioff |
VT (V) |
| Cytop dielectric. PMMA dielectric. μavg indicates the average mobility of 6 devices. |
| Ph(DPPT2)2a |
(8.5 ± 0.5) × 10−2 |
0.090 |
>103 |
22.4 |
| Ph(DPPT2)2b |
0.113 ± 0.003 |
0.115 |
>104 |
2.6 |
| NA(DPPT2)2a |
(3.70 ± 0.27) × 10−2 |
0.039 |
>103 |
23.6 |
| Py(DPPT2)2a |
(6.47 ± 0.13) × 10−4 |
6.48 × 10−4 |
>103 |
8.9 |
We also tried fabricating top-gate, bottom-contact devices with PMMA (diluted in n-butyl acetate) which is commonly used dielectrics in OFETs. All other processing parameters for the PMMA-based devices were the same as the Cytop-based devices. However, at this condition, the devices based on NA(DPPT2)2 and Py(DPPT2)2 could not be obtained because the n-butyl acetate solvent for PMMA would dissolve the organic semiconducting materials of NA(DPPT2)2 and Py(DPPT2)2. Only Ph(DPPT2)2 FETs were successfully fabricated with PMMA as the dielectric. Fig. 10 shows the electrical transfer characteristics of the devices, and the performance of the devices is summarized in Table 2. It is noted that the PMMA-based devices exhibited better performance than Cytop-based devices. The hole mobility of Ph(DPPT2)2 in the PMMA devices can be as high as 0.12 cm2 V−1 s−1. It is expected that higher mobilities could be achieved by further optimizing the device fabrication process, such as the selection of proper annealing temperature above the Tc of Ph(DPPT2)2 or the chosen of appropriate dielectrics, etc.
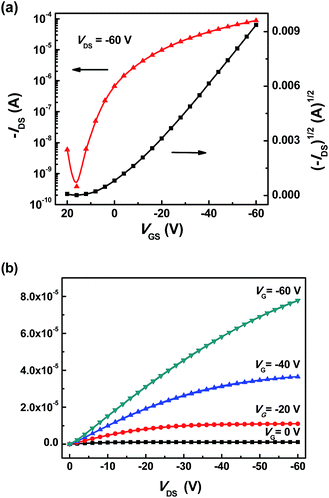 |
| | Fig. 10 (a) Typical transfer and (b) output plot (VDS = −60 V) of organic field-effect transistors annealed at 120 °C from spin-casted Ph(DPPT2)2, the device with PMMA as dielectrics. | |
Conclusions
We have synthesized four novel D–A–π–A–D small molecules of Ar(DPPT2)2, incorporating different π-bridge aryl units (phenyl, naphthyl, anthryl and pyridyl) via a Pd-catalyzed, direct C–H arylation reaction. We have found that the nature of the core π-bridge aryl unit has a significant effect on the optoelectronic properties and OFET performance. The new DPP derivatives afforded low band gaps and low-lying HOMOs as indicated by optical spectroscopy and cyclic voltammetry. Hole mobilities as high as 0.12 cm2 V−1 s−1 for Ph(DPPT2)2 with Ion/Ioff greater than 104 have been measured even under ambient conditions. It is found that for the similar molecular backbone, π-bridged aryl units like phenyl is more preferred for efficient hole transport than the bulky naphthyl and anthryl as well as the electron-withdrawing pyridyl. Our work sheds more light onto the understanding of the correlation between the molecular structure and charge transport properties of organic semiconductors.
Acknowledgements
The authors thank the National Natural Science Foundation of China (21304047), NSF of Jiangsu Province (BK20130919 and 13KJB430017) and Research Fund for the Doctoral Program of Higher Education (20133221120015). K. B. gratefully acknowledges financial support from the German Research Foundation (BR 4869 1-1) and the Diamond Light Source. J. N. acknowledges support from the Diamond Light Source and from the project CEITEC 2020 (grant No. LQ1601 financed by the MEYS of the Czech Republic). We would like to thank Dr T. Arnold (Diamond Light Source), D. Harkin (Univ. of Cambridge), and J. Rozbořil (Masaryk University) for support during synchrotron experiment.
Notes and references
- L. Bürgi, M. Turbiez, R. Pfeiffer, F. Bienewald, H.-J. Kirner and C. Winnewisser, Adv. Mater., 2008, 20, 2217–2224 CrossRef.
- A. B. Tamayo, M. Tantiwiwat, B. Walker and T.-Q. Nguyen, J. Phys. Chem. C, 2008, 112, 15543–15552 CAS.
- H. Chen, Y. Guo, G. Yu, Y. Zhao, J. Zhang, D. Gao, H. Liu and Y. Liu, Adv. Mater., 2012, 24, 4618–4622 CrossRef CAS PubMed.
- C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859–1880 CrossRef CAS PubMed.
- I. Kang, T. K. An, J.-a. Hong, H.-J. Yun, R. Kim, D. S. Chung, C. E. Park, Y.-H. Kim and S.-K. Kwon, Adv. Mater., 2013, 25, 524–528 CrossRef CAS PubMed.
- Y. Li, P. Sonar, L. Murphy and W. Hong, Energy Environ. Sci., 2013, 6, 1684–1710 CAS.
- M. Kaur and D. H. Choi, Chem. Soc. Rev., 2015, 44, 58–77 RSC.
- X. Guo, S. R. Puniredd, M. Baumgarten, W. Pisula and K. Müllen, Adv. Mater., 2013, 25, 5467–5472 CrossRef CAS PubMed.
-
(a) J. Yao, C. Yu, Z. Liu, H. Luo, Y. Yang, G. Zhang and D. Zhang, J. Am. Chem. Soc., 2016, 138, 173–185 CrossRef CAS PubMed;
(b) J. Li, Y. Zhao, H. S. Tan, Y. Guo, C. A. Di, G. Yu, Y. Liu, M. Lin, S. H. Lim, Y. Zhou, H. Su and B. S. Ong, Sci. Rep., 2012, 2, 754 Search PubMed.
- C. Kanimozhi, N. Yaacobi-Gross, K. W. Chou, A. Amassian, T. D. Anthopoulos and S. Patil, J. Am. Chem. Soc., 2012, 134, 16532–16535 CrossRef CAS PubMed.
- I. Kang, H.-J. Yun, D. S. Chung, S.-K. Kwon and Y.-H. Kim, J. Am. Chem. Soc., 2013, 135, 14896–14899 CrossRef CAS PubMed.
- H.-J. Yun, S.-J. Kang, Y. Xu, S. O. Kim, Y.-H. Kim, Y.-Y. Noh and S.-K. Kwon, Adv. Mater., 2014, 26, 7300–7307 CrossRef CAS PubMed.
- A. R. Han, G. K. Dutta, J. Lee, H. R. Lee, S. M. Lee, H. Ahn, T. J. Shin, J. H. Oh and C. Yang, Adv. Funct. Mater., 2015, 25, 247–254 CrossRef CAS.
- M. Singh, R. Kurchania, J. A. Mikroyannidis, S. S. Sharma and G. D. Sharma, J. Mater. Chem. A, 2013, 1, 2297–2304 CAS.
- J. Roncali, P. Leriche and P. Blanchard, Adv. Mater., 2014, 26, 3821–3838 CrossRef CAS PubMed.
- B. Walker, C. Kim and T. Q. Nguyen, Chem. Mater., 2011, 23, 470–482 CrossRef CAS.
-
(a) H. Zhong, J. Smith, S. Rossbauer, A. J. P. White, T. D. Anthopoulos and M. Heeney, Adv. Mater., 2012, 24, 3205–3211 CrossRef CAS PubMed;
(b) C. Lu and W. C. Chen, Chem.–Asian J., 2013, 8, 2813–2821 CrossRef CAS PubMed.
- K. A. Mazzio, M. Yuan, K. Okamoto and C. K. Luscombe, ACS Appl. Mater. Interfaces, 2011, 3, 271–278 CAS.
- S. Loser, C. J. Bruns, H. Miyauchi, R. P. Ortiz, A. Facchetti, S. I. Stupp and T. J. Marks, J. Am. Chem. Soc., 2011, 133, 8142–8145 CrossRef CAS PubMed.
- C. Wang, Y. Zang, Y. Qin, Q. Zhang, Y. Sun, C.-A. Di, W. Xu and D. Zhu, Chem.–Eur. J., 2014, 20, 13755–13761 CrossRef CAS PubMed.
- S. L. Suraru, U. Zschieschang, H. Klauk and F. Wurthner, Chem. Commun., 2011, 47, 1767–1769 RSC.
- P.-L. T. Boudreault, J. W. Hennek, S. Loser, R. P. Ortiz, B. J. Eckstein, A. Facchetti and T. J. Marks, Chem. Mater., 2012, 24, 2929–2942 CrossRef CAS.
- Z. Cai, H. Luo, X. Chen, G. Zhang, Z. Liu and D. Zhang, Chem.–Asian J., 2014, 9, 1068–1075 CrossRef CAS PubMed.
- Y. Qiao, Y. Guo, C. Yu, F. Zhang, W. Xu, Y. Liu and D. Zhu, J. Am. Chem. Soc., 2012, 134, 4084–4087 CrossRef CAS PubMed.
- Y. Zhang, C. Kim, J. Lin and T.-Q. Nguyen, Adv. Funct. Mater., 2012, 22, 97–105 CrossRef CAS.
- W. S. Yoon, S. K. Park, I. Cho, J.-A. Oh, J. H. Kim and S. Y. Park, Adv. Funct. Mater., 2013, 23, 3519–3524 CrossRef CAS.
- H. Zhong, J. Smith, S. Rossbauer, A. J. P. White, T. D. Anthopoulos and M. Heeney, Adv. Mater., 2012, 24, 3205–3211 CrossRef CAS PubMed.
- A. B. Tamayo, B. Walker and T.-Q. Nguyen, J. Phys. Chem. C, 2008, 112, 11545–11551 CAS.
- B. Walker, A. B. Tamayo, X.-D. Dang, P. Zalar, J. H. Seo, A. Garcia, M. Tantiwiwat and T.-Q. Nguyen, Adv. Funct. Mater., 2009, 19, 3063–3069 CrossRef CAS.
- J. Huang, C. Zhan, X. Zhang, Y. Zhao, Z. Lu, H. Jia, B. Jiang, J. Ye, S. Zhang, A. Tang, Y. Liu, Q. Pei and J. Yao, ACS Appl. Mater. Interfaces, 2013, 5, 2033–2039 CAS.
- J.-Y. Pan, L.-J. Zuo, X.-L. Hu, W.-F. Fu, M.-R. Chen, L. Fu, X. Gu, H.-Q. Shi, M.-M. Shi, H.-Y. Li and H.-Z. Chen, ACS Appl. Mater. Interfaces, 2013, 5, 972–980 CAS.
- M. Chen, W. Fu, M. Shi, X. Hu, J. Pan, J. Ling, H. Li and H. Chen, J. Mater. Chem. A, 2013, 1, 105–111 CAS.
- J. Liu, B. Walker, A. Tamayo, Y. Zhang and T.-Q. Nguyen, Adv. Funct. Mater., 2013, 23, 47–56 CrossRef CAS.
- P. Sonar, G.-M. Ng, T. T. Lin, A. Dodabalapur and Z.-K. Chen, J. Mater. Chem., 2010, 20, 3626–3636 RSC.
- B. Walker, J. Liu, C. Kim, G. C. Welch, J. K. Park, J. Lin, P. Zalar, C. M. Proctor, J. H. Seo, G. C. Bazan and T.-Q. Nguyen, Energy Environ. Sci., 2013, 6, 952–962 CAS.
- Y. Z. Lin, P. Cheng, Y. F. Li and X. W. Zhan, Chem. Commun., 2012, 48, 4773–4775 RSC.
- O. P. Lee, A. T. Yiu, P. M. Beaujuge, C. H. Woo, T. W. Holcombe, J. E. Millstone, J. D. Douglas, M. S. Chen and J. M. Frechet, Adv. Mater., 2011, 23, 5359–5363 CrossRef CAS PubMed.
- J. Liu, Y. Sun, P. Moonsin, M. Kuik, C. M. Proctor, J. Lin, B. B. Hsu, V. Promarak, A. J. Heeger and T.-Q. Nguyen, Adv. Mater., 2013, 25, 5898–5903 CrossRef CAS PubMed.
- J. W. Jung, T. P. Russell and W. H. Jo, Chem. Mater., 2015, 27, 4865–4870 CrossRef CAS.
- Y. S. Choi and W. H. Jo, Org. Electron., 2013, 14, 1621–1628 CrossRef CAS.
- Y. Lin, H. F. Dam, T. R. Andersen, E. Bundgaard, W. Fu, H. Chen, F. C. Krebs and X. Zhan, J. Mater. Chem. C, 2013, 1, 8007 RSC.
- H. Bai, P. Cheng, Y. Wang, L. Ma, Y. Li, D. Zhu and X. Zhan, J. Mater. Chem. A, 2014, 2, 778–784 CAS.
- Y. Zhang, J. Liu and T.-Q. Nguyen, ACS Appl. Mater. Interfaces, 2013, 5, 2347–2353 CAS.
- Y. Lin, Y. Li and X. Zhan, Adv. Energy Mater., 2013, 3, 724–728 CrossRef CAS.
- Y. Lin, L. Ma, Y. Li, Y. Liu, D. Zhu and X. Zhan, Adv. Energy Mater., 2013, 3, 1166–1170 CrossRef CAS.
- H. Burckstummer, A. Weissenstein, D. Bialas and F. Wurthner, J. Org. Chem., 2011, 76, 2426–2432 CrossRef PubMed.
- J. W. Jung and W. H. Jo, Chem. Mater., 2015, 27, 6038–6043 CrossRef CAS.
- X. Duan, M. Xiao, J. Chen, X. Wang, W. Peng, L. Duan, H. Tan, G. Lei, R. Yang and W. Zhu, ACS Appl. Mater. Interfaces, 2015, 7, 18292–18299 CAS.
- X.-F. Wu, W.-F. Fu, Z. Xu, M. Shi, F. Liu, H.-Z. Chen, J.-H. Wan and T. P. Russell, Adv. Funct. Mater., 2015, 25, 5954–5966 CrossRef CAS.
- L. G. Mercier and M. Leclerc, Acc. Chem. Res., 2013, 46, 1597–1605 CrossRef CAS PubMed.
- M. Lafrance, C. N. Rowley, T. K. Woo and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 8754–8756 CrossRef CAS PubMed.
- M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496–16497 CrossRef CAS PubMed.
- Q. Guo, J. Dong, D. Wan, D. Wu and J. You, Macromol. Rapid Commun., 2013, 34, 522–527 CrossRef CAS PubMed.
-
(a) J.-R. Pouliot, B. Sun, M. Leduc, A. Najari, Y. Li and M. Leclerc, Polym. Chem., 2015, 6, 278–282 RSC;
(b) A. K. Palai, A. Kumar, K. Sim, J. Kwon, T. J. Shin, S. Jang, S. Cho, S.-U. Park and S. Pyo, New J. Chem., 2016, 40, 385–392 RSC.
- S.-Y. Liu, M.-M. Shi, J.-C. Huang, Z.-N. Jin, X.-L. Hu, J.-Y. Pan, H.-Y. Li, A. K. Y. Jen and H.-Z. Chen, J. Mater. Chem. A, 2013, 1, 2795–2805 CAS.
- S. Y. Liu, W. Q. Liu, J. Q. Xu, C. C. Fan, W. F. Fu, J. Ling, J. Y. Wu, M. M. Shi, A. K. Jen and H. Z. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 6765–6775 CAS.
-
(a) A. D. Hendsbee, J.-P. Sun, L. R. Rutledge, I. G. Hill and G. C. Welch, J. Mater. Chem. A, 2014, 2, 4198 RSC;
(b) X.-C. Li, H. Sirringhaus, F. Garnier, A. B. Holmes, S. C. Moratti, N. Feeder, W. Clegg, S. J. Teat and R. H. Friend, J. Am. Chem. Soc., 1998, 120, 2206–2207 CrossRef CAS.
- J. H. Gao, R. J. Li, L. Q. Li, Q. Meng, H. Jiang, H. X. Li and W. P. Hu, Adv. Mater., 2007, 19, 3008–3011 CrossRef CAS.
- M. Jung, Y. Yoon, J. Park, W. Cha, A. Kim, J. Kang, S. Gautam, D. Seo, J. Cho, H. Kim, J. Choi, K. Chae, K. Kwak, H. Son, M. Ko, H. Kim, D. Lee, J. Kim, D. Choi and B. Kim, ACS Nano, 2014, 8, 5988–6003 CrossRef CAS PubMed.
- S. Wang, J. Yang, Z. Zhang, Y. Hu, X. Cao, H. Li, Y. Tao, Y. Li and W. Huang, RSC Adv., 2015, 5, 68192–68199 RSC.
-
(a) M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef;
(b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- Y. Sun, G. Welch, W. L. Leong, C. J. Takacs, G. C. Bazan and A. J. Heeger, Nat. Mater., 2012, 11, 44–48 CrossRef CAS PubMed.
- J. H. Park, E. H. Jung, J. W. Jung and W. H. Jo, Adv. Mater., 2013, 25, 2583–2588 CrossRef CAS PubMed.
- S. K. Lee, J. M. Cho, Y. Goo, W. S. Shin, J. Lee, W. Lee, I. Kang, H. Shim and S. Moon, Chem. Commun., 2011, 47, 1791–1793 RSC.
- M. M. Wienk, M. Turbiez, J. Gilot and R. A. Janssen, Adv. Mater., 2008, 20, 2556–2560 CrossRef CAS.
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Acc. Chem. Res., 2010, 43, 1396–1407 CrossRef CAS PubMed.
-
(a) T. Kono, D. Kumaki, J. Nishida, S. Tokito and Y. Yamashita, Chem. Commun., 2010, 46, 3265–3267 RSC;
(b) A. Irfan, M. Nadeem, M. Athar, F. Kanwal and J. Zhang, Comput. Theor. Chem., 2011, 968, 8–11 CrossRef CAS;
(c) C. R. Newman, C. D. Frisbie, D. A. da Silva Filho, J.-L. Brédas, P. C. Ewbank and K. R. Mann, Chem. Mater., 2004, 16, 4436–4451 CrossRef CAS.
- W. Kang, M. Jung, W. Cha, S. Jang, Y. Yoon, H. Kim, H. J. Son, D.-K. Lee, B. Kim and J. H. Cho, ACS Appl. Mater. Interfaces, 2014, 6, 6589–6597 CAS.
- Z. Liu, J. H. Oh, M. E. Roberts, P. Wei, B. C. Paul, M. Okajima, Y. Nishi and Z. Bao, Appl. Phys. Lett., 2009, 94, 203301 CrossRef.
- C. J. Mueller, C. R. Singh, M. Fried, S. Huettner and M. Thelakkat, Adv. Funct. Mater., 2015, 25, 2725–2736 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra10832f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 










