DOI:
10.1039/C6RA10641B
(Paper)
RSC Adv., 2016,
6, 57183-57189
POSS-based meso-/macroporous covalent networks: supporting and stabilizing Pd for Suzuki–Miyaura reaction at room temperature†
Received
25th April 2016
, Accepted 30th May 2016
First published on 31st May 2016
Abstract
A facile template-free strategy for the fabrication of organic–inorganic meso-/macroporous covalent networks was reported by the combination of the aminopropyl-polyhedral oligomeric silsesquioxanes (POSS) with terephthalic aldehyde (TPA) through Schiff base chemistry reactions. Such porous covalent networks have high BET surface areas and various abundant nitrogen-containing functionalities that could serve as both supports and stabilizers for homogeneous palladium (Pd) catalyst. The resulting heterogeneous Pd catalyst (Pd/POSS–TPAa) exhibited enhanced catalytic activity for the Suzuki–Miyaura reactions at room temperature with a short reaction time, and the catalyst can be easily recycled up to five times without significant loss in the conversion and the leaching of Pd. The excellent performance of Pd/POSS–TPAa is mostly attributed to the strong coordination between N-containing groups in the covalent framework and Pd.
Introduction
Over the past few decades, design and fabrication of nanoporous materials, including porous silicas,1 porous carbons,2 porous metal oxides,3 metal–organic frameworks,4 porous organic polymers,5 and organic–inorganic porous hybrids,6 have attracted increasing attention due to their versatile structures, high surface area, and tunable surface chemistry, which allow potential applications in gas storage, explosive detection, drug release, and catalysis. Although significant progress has been achieved in structural, compositional, and topological control over polymeric framework and pore structure in materials field,7 their existing synthesis methods, such as hydrolytic polymerization,8 nucleophilic reaction,9 ring-opening polymerization,10 and radical polymerization11 are energy-consuming, time-demanding, and procedure tedious. Additionally, such porous materials are usually linked through Si–O–Si, C–C, or C–O single bonds, and the introduction of multi-functionalities represents another significant challenge. Hence, developing an easy-to-make and cost-effective process for the synthesis of functional porous materials is highly desirable.
Palladium (Pd)-catalyzed Suzuki–Miyaura (SM) reactions, recognized by the award of the Nobel Prize in Chemistry in 2010, are of strategic importance in forming carbon–carbon bonds in synthetic chemistry and industrial applications.12 However, the SM reaction was usually performed under homogeneous conditions, and the recovery of costly, unstable and potentially toxic Pd catalysts remains as a pressing issue. The immobilization or entrapping of Pd on various porous supports is a commonly used method to prepare heterogeneous Pd catalysts.13 Nevertheless, most of the obtained heterogeneous Pd catalysts often result in low activities and leaching of active sites because of the weak interaction between Pd and supports. Additionally, due to the high activation barrier of substrates associated with the rate-limiting steps, harsh reaction conditions such as high temperatures or long reaction time are still undesirable. Actually, the functional support, in many cases, is not a mere inert spectator used for the catalyst heterogenization, it can play an active role in tuning the catalytic activity and stability.14 Therefore, the rational design and synthesis of a porous support that not only have strong interaction with Pd but also could significantly increase the efficiency of the heterogeneous Pd catalysts for liquid phase SM reactions under mild conditions is of great interest.
Schiff base chemistry reaction, a versatile tool in organic synthesis based on covalent bonding amino and carbonyl groups, recently has been successfully utilized in the preparation of porous polymers.15 These polymers have received considerable interest as metal catalyst supports due to their abundant nitrogen (N)-containing groups, which are helpful in improving the dispersity, stability, and the catalytic performance of metal catalysts.16 Although Schiff base chemistry has demonstrated to be a promising approach for the preparation of porous materials owing to the dynamic nature of imine bond formation, the current studies have thus far been mostly devoted to covalent–organic frameworks (COFs). In this study, we explore for the first time a new strategy for a template-free preparation of N-rich organic–inorganic porous covalent networks with high surface area through Schiff base chemistry reaction. By accessible coordination with nitrogen groups on porous framework, such an exceptional family of hybrid materials could serve as both stabilizers and supports for Pd catalyst. And the resultant heterogeneous Pd catalyst exhibited excellent activity and stability for the SM reactions at room temperature with a short reaction time.
Experimental details
Materials and analysis
All chemicals were used as received without further purification. FT-IR spectra were recorded on a Nicolet 360 FT-IR instrument (KBr discs) in the 4000–400 cm−1 region. CHN elemental analysis was carried out on an elemental analyzer Vario EL cube. ICP-AES was performed by a P-4010 (Hitachi, Japan) spectrometer. TG analysis was performed with a STA409 instrument in dry air at a heating rate of 10 °C min−1 from 100 to 800 °C. The nitrogen sorption isotherms and pore size distribution curves were measured at the temperature of liquid nitrogen (77 K) using a BELSORP-MINI analyzer. Scanning electron microscopy (SEM) images were recorded on a SUPERSCAN SSX-550 electron microscope (Shimadz, Japan) operating at 20 kV. Field emission scanning electron microscope (FESEM; Hitachi S-4800, accelerated voltage: 5 kV) accompanied by Energy Dispersive X-ray spectrometry (EDS; accelerated voltage: 20 kV) was used to study the morphology and the elements distribution. TEM images were obtained with JEOL JEM-2100 electron microscope operated at 200 kV. X-ray photoelectron spectroscopy (XPS) was conducted on a PHI 5000 Versa Probe Xray photoelectron spectrometer equipped with Al Kα radiation (1486.6 eV). The C 1s peak at 285.2 eV was used as the reference for the binding energies. XRD patterns were collected on the Bruker D8 Advance powder diffractometer using Ni-filtered Cu/Kα radiation source at 40 kV and 20 mA, from 5 to 60° with a scan rate of 4° min−1.
Catalyst preparation
Synthesis of octa(aminopropyl silsesquioxane) (POSS). Typically, deionized water (18 mL), propyl alcohol (8 mL), acetonitrile (2 mL), and tetraethyl ammonium hydroxide (0.4 mL) were added into a 500 mL flask to get a heterogeneous solution. γ-Aminopropyl triethoxysilane (44.0 g) was added into the solution, and the obtained mixture was heated to 50 °C with vigorous stirring. After reaction 12 h, the formed white crystalline precipitate product POSS was filtered and washed with cold methanol for three times, followed by drying in vacuum at 80 °C for 24 h.
Synthesis of POSS–TPAx. In a 100 mL round-bottom flask, POSS (0.5 g, 5.68 mmol) was dissolved in a mixture solvent of dimethylformamide (DMF, 10 mL) and deionized water (H2O, 3 mL) to form a homogeneous, transparent solution, and the solution was heated to 100 °C. When the DMF solution (5 mL) containing terephthalic aldehyde (TPA, 0.075 g, 5.68 mmol) was added into the above solution, white precipitates formed immediately. The mixture was vigorously stirred at 100 °C for 12 h. Then, the precipitates were collected by filtration, washed with ethanol for three times, and dried at 50 °C under vacuum for 24 h to afford POSS–TPAa. The samples POSS–TPAb, POSS–TPAc, and POSS–TPAd were prepared accordingly using the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 3:1, and 4:1 molar ratio of POSS to TPA, respectively.
:1, 3:1, and 4:1 molar ratio of POSS to TPA, respectively.
Synthesis of POSS–TPAa–BA. The POSS–TPAa (200 mg) was dispersed in 10 mL of dry methanol, and benzaldehyde (BA, 130 mg) was added dropwise into it. The dispersion was stirred vigorously at 65 °C for 12 h. The product POSS–TPAa–BA were isolated by centrifugation, washed with methanol for three times and dried under vacuum overnight at 70 °C.
Synthesis of Pd/POSS–TPAx. The POSS–TPAa (200 mg) was stirred with dichloromethane (10 mL) in a 25 mL round bottom flask. Palladium acetate (20 mg) was added into the above solution, and the mixture was stirred at 50 °C for 24 h. The solids Pd/POSS–TPAa were separated via centrifugation, washed thoroughly with deionized water and ethanol, and finally dried overnight under vacuum. Pd/POSS–TPAd and Pd/POSS–TPAa–BA were prepared following the same procedures as for Pd/POSS–TPAa.
Catalytic test
Typically, in a 25 mL round-bottom flask, phenylboronic acid (1.5 mmol), bromobenzene (1 mmol), Na2CO3 (0.15 g), and catalyst Pd/POSS–TPAa (0.02 g, 0.0036 mmol based on Pd) were added into the mixed solvent (3 mL deionized water and 3 mL ethanol). The resulting mixture was reacted at room temperature for 30 min. After completion of the reaction, the reaction mixture was filtered and the filtrate was analyzed by gas chromatography (GC, SP-6890A) equipped with a FID detector and a capillary column (SE-54 30 m × 0.32 mm × 0.25 μm). The catalyst was recovered by filtration and washed with ethanol and deionized water, respectively, then dried in vacuum to provide the recovered catalyst, and finally reused in the next run without addition of any fresh catalyst.
Results and discussion
Preparation and characterization of catalysts
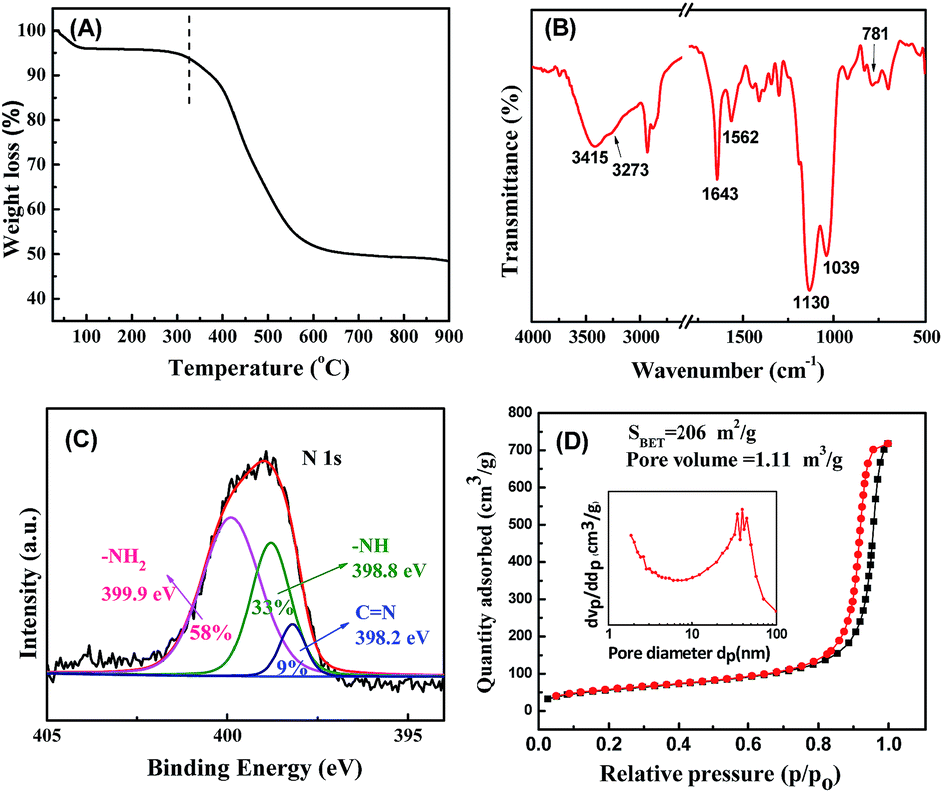
In view of the relevance of Schiff base chemistry and the search for N-rich polymers in porous material science, POSS,10,17 an intriguing class of organic–inorganic hybrid materials, possess a cubic structure represented by the formula R8Si8O12, has been selected as the building blocks to reacted with TPA. The synthetic process is schematically illustrated in Fig. 1. POSS (see XRD in Fig. S1 in ESI†) was dissolved in a mixture solvent of dimethylformamide (DMF) and deionized water (H2O) to form a homogeneous, transparent solution. When the DMF solution containing TPA was added into the above solution, white precipitates formed immediately. This is because the amino groups (–NH2) on POSS were reacted with aldehyde groups (–CHO) on TPA via Schiff base reaction to yield insoluble cross-linked polymer networks.18 The precipitates in 80–85% high yield were collected by filtration and dried at 50 °C under vacuum for 24 h, denoted as POSS–TPAa. CHN elemental analysis indicates that the actual molar ratio of TPA to POSS in POSS–TPAa is about 2.8:1 (Table S1 in ESI†), and POSS–TPAa was found to be stable thermally up to ca. 350 °C as evidenced by thermogravimetric analysis (TG) (Fig. 2A).
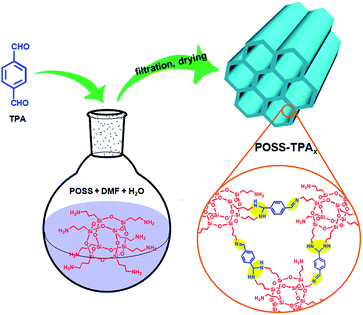 |
| | Fig. 1 Schematic illustration outlining the preparation and structure of POSS–TPAx. | |
 |
| | Fig. 2 (A) TG curve, (B) FTIR spectrum, (C) XPS N 1s spectrum, and (D) nitrogen adsorption–desorption isotherms of POSS–TPAa. | |
Fig. 2B illustrates the FTIR spectrum of POSS–TPAa. The distinct bands corresponding to the Si–O–Si (1130 cm−1) and aromatic ring stretching vibrations (1300–1550 cm−1), as well as to the primary amine group –NH2 at 3415 cm−1 (stretching vibration) and 1562 cm−1 (deformation vibration) are present in the FT-IR spectrum, indicating the presence POSS and TPA building blocks. Meanwhile, POSS–TPAa shows the moderate intensity absorption band at 1643 cm−1 assigned to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and the bands at 3273 and 781 cm−1 attributed to the stretching vibration and bending vibration of secondary amine N–H, respectively, suggesting the successful combination of POSS and TPA through Schiff base reaction. Furthermore, the high resolution N 1s spectrum of POSS–TPAa analyzed by X-ray photo-electron spectroscopy (XPS) is shown in Fig. 2C. The dominant peak at 399.9 eV was ascribed to the terminal –NH2, and the peaks at 398.8 eV and 398.2 eV were attributed to the –NH and CN, respectively, which gives a –NH2/–NH/CN ratio of 58:38:9. This XPS result also confirms the successful build-up of a covalent network for POSS–TPAa through Schiff base reaction.
N and the bands at 3273 and 781 cm−1 attributed to the stretching vibration and bending vibration of secondary amine N–H, respectively, suggesting the successful combination of POSS and TPA through Schiff base reaction. Furthermore, the high resolution N 1s spectrum of POSS–TPAa analyzed by X-ray photo-electron spectroscopy (XPS) is shown in Fig. 2C. The dominant peak at 399.9 eV was ascribed to the terminal –NH2, and the peaks at 398.8 eV and 398.2 eV were attributed to the –NH and CN, respectively, which gives a –NH2/–NH/CN ratio of 58:38:9. This XPS result also confirms the successful build-up of a covalent network for POSS–TPAa through Schiff base reaction.
The porous structure of POSS–TPAa was then characterized by nitrogen sorption analysis at 77 K. The N2 adsorption–desorption isotherms in Fig. 2D display a typical IV type isotherm with an apparent H1-type hysteresis loop characteristic of mesoporous structure as well as macroporous. The corresponding pore size distribution of POSS–TPAa is between 10 and 100 nm. The specific Brunauer–Emmett–Teller (BET) surface area and the pore volume were calculated to be 206 m2 g−1 and 1.11 cm3 g−1, respectively. SEM characterization (Fig. 3A) reveals the distinctive 3D skeletal morphology of the meso-/macroporous POSS–TPAa, and the EDS mapping (Fig. S2 in ESI†) image validates the homogeneous distribution of Si element in the porous polymer frameworks.
 |
| | Fig. 3 SEM images of (A) POSS–TPAa, (B) POSS–TPAb, (C) POSS–TPAc, and (D) POSS–TPAd. | |
A series POSS–TPAx (x = a, b, c, d) covalent networks (see FTIR and TG characterizations in Fig. S3 and S4 in ESI†) were synthesized by changing the molar ratio of two precursors POSS and TPA. It was found that the precursor molar ratio of TPA/POSS has less effects for the POSS–TPA′x composition, as the TPA/POSS molar ratio increases from 1:1 to 3:1, the actual ratio of TPA/POSS in obtained samples POSS–TPAa, POSS–TPAb, and POSS–TPAc are maintained 1:2.7–1:2.9, as demonstrated by CNH elemental analysis (Table S1 in ESI†). A relative high molar ratio of 3.3:1 in POSS–TPAd could be achieved when using a rather high TPA/POSS precursor ratio of 4:1. Nitrogen adsorption–desorption isotherms for all these samples reveal the meso-/macroporous structure (Fig. S5 in ESI†). However, the BET surface areas tend to decrease as the TPA/POSS precursor ratio increased (Fig. 3), and 206, 151.6, 143.7, and 120.1 m2 g−1 surface areas were observed for POSS–TPAa, POSS–TPAb, and POSS–TPAc, and POSS–TPAd, respectively. This is presumably because the superfluous TPA degenerated the order of mesostructure and increased the cross-link degree of the covalent network. These variations could also be distinguished from the SEM images in Fig. 3, the 3D skeletal morphology with macroporous or mesoporous pore size on the surface for POSS–TPAx is becoming more and more not obvious when the x changed form a to d. Particularly, POSS–TPAd displays less porous on the surface (Fig. 3D).
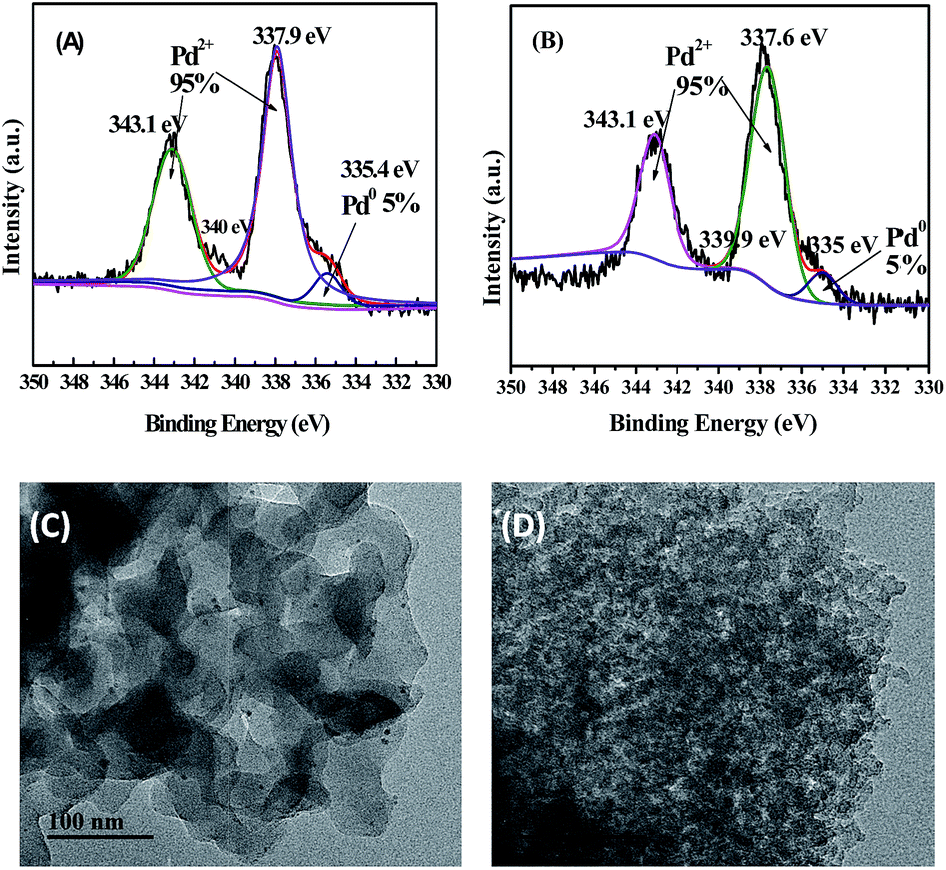
Obviously, these cross-linked porous covalent networks, with various abundant N-containing groups, indeed create a regiospecific chemical environment for the variability and immobilization of homogeneous metal catalysts. Subsequently, two typical supported Pd catalysts Pd/POSS–TPAa and Pd/POSS–TPAd were prepared by immersion POSS–TPAa and POSS–TPAd in Pd(OAc)2 dichloromethane solution, respectively, followed by washing and drying. About 2 wt% Pd content in the obtained catalysts was determined by inductively coupled plasma (ICP) (Table 1). The solid catalysts Pd/POSS–TPAa and Pd/POSS–TPAd were analysed by XPS. The Pd 3d3/2, 3d5/2 binding energy in Fig. 4A and B demonstrate that the metal ligated to porous covalent network supports corresponds to Pd2+ (343.1 eV for 3d5/2 and 337.9 eV for 3d3/2) with a fraction of the reduced Pd0 (335.4 eV for 3d3/2). This indicates that strong coordination of N-to-Pd is formed within the porous covalent networks. Fig. 4C and D present the TEM images of Pd/POSS–TPAa and Pd/POSS–TPAd, respectively. The observed porous structures are in consistent well with those of the SEM images. In the TEM image of Pd/POSS–TPAa, a small amount of Pd nanoparticles with quite small size of 2–3 nm could be seen, but it is not obvious in the TEM image of Pd/POSS–TPAd due to the high-density of dark micelles. EDS elemental mapping image shows abundant Pd elements that are uniformly dispersed in the porous covalent networks (Fig. S6 in ESI†).
Table 1 Suzuki coupling of bromobenzene with phenylboronic acid by various catalystsa
| Entry |
Catalyst |
Pd (wt%) |
Yieldb (%) |
TOFc |
| 10 min |
30 min |
| Reaction conditions: phenylboronic acid (1.5 mmol), bromobenzene (1 mmol), Na2CO3 (0.15 g), deionized water (3 mL) and ethanol (3 mL), catalyst amount (0.02 g, 0.0036 mmol based on Pd), room temperature. The yield of product. TOF = [mol product]/([mol Pd][reaction time 1/6 h]). |
| 1 |
Pd/POSS–TPAa |
1.93 |
92 |
99 |
1533 |
| 2 |
Pd/POSS–TPAd |
2.04 |
41 |
46 |
683 |
| 3 |
Pd/POSS–TPAa–BA |
1.97 |
35 |
40 |
583 |
 |
| | Fig. 4 XPS spectra of Pd 3d for (A) Pd/POSS–TPAa and (B) Pd/POSS–TPAd; TEM images of (C) Pd/POSS–TPAa and (D) Pd/POSS–TPAd. | |
Catalytic activity for the Suzuki coupling reactions
Catalytic tests were first performed for the coupling of bromobenzene with phenylboronic acid using the present solid Pd catalysts at room temperature, and the results are shown in Table 1. Pd/POSS–TPAa exhibited a high yield of 92% within only 10 min, and the TOF was found to be 1533 per mole catalyst per hour (entry 1). With 0.5 h reaction time, the excellent 99% yield could be achieved without an observation of any side products. The influence of catalyst amount and the molar ratio of phenylboronic acid to bromobenzene on the yield of cyclic carbonate was investigated, and the results are compiled in Tables S2 and S3 in ESI.† With only 0.01 g (0.0018 mmol Pd) catalyst, 90% yield could be obtained with a very high TOF of 3033 per mole catalyst per hour. However, Pd/POSS–TPAd offered a low yield of 46% with 0.5 h (entry 2). The catalytic activities of these catalysts are reasonably related to their BET surface areas, the comparatively high surface area favour higher catalytic activity. It seems that the difference of the specific surface areas between Pd/POSS–TPAa and Pd/POSS–TPAd is difficult to have such significant effects on the catalytic activity. On the other hand, as it is well known in the literature, the functional N-containing groups usually play important roles for Pd catalysts in aqueous catalysis.16 We thus consider that how the N-containing groups influences the catalytic activity and if there is some difference between the supports POSS–TPAa and POSS–TPAd. To address these issues, POSS–TPAd was also analyzed by XPS. The high resolution N 1s spectrum in Fig. 5 displays a 27:64:9 molar ratio of –NH2/–NH/CN. Compared with 58:38:9 in POSS–TPAa, it can be seen that POSS–TPAa has a much higher –NH2 content than that of POSS–TPAd. It is thus proposed that the –NH2 groups on the covalent framework play promotional effects for Pd catalysts probably attribute to the coordination and electron transfer from nitrogen to Pd.19 In order to further verify this conclusion, the catalyst Pd/POSS–TPAa–BA, in which benzaldehyde (BA) was anchored to the POSS–TPAa through Schiff base reaction with the rest –NH2 groups, was prepared and used as a control catalyst for the coupling of bromobenzene with phenylboronic acid. In contrast to Pd/POSS–TPAa, Pd/POSS–TPAa–BA led to a very low yield of 40% (entry 3). It was even more active than that of Pd/POSS–TPAd. This result further confirms that the –NH2 groups play promotional effects for Pd catalysts in SM coupling reaction. We then further tested the feasibility of Pd/POSS–TPAa as catalyst for a series of Suzuki coupling reactions (Table 2). It can be seen that good to excellent yields were obtained almost in all cases at room temperature. Not only can our new catalyst Pd/POSS–TPAa be comparable to the previously reported Pd catalysts in yield and selectivity, but also the TOF is much higher (Table S4 in ESI†).
 |
| | Fig. 5 XPS N 1s spectrum of Pd/POSS–TPAd. | |
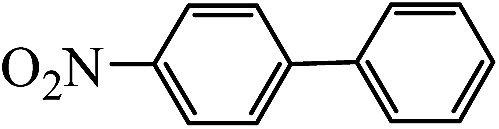
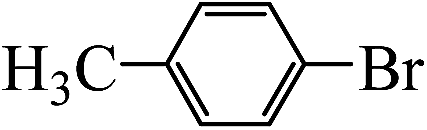
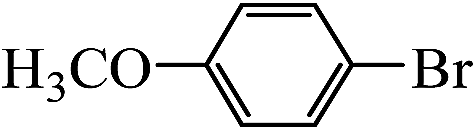
Table 2 Suzuki coupling of various phenylboronic acids with aryl halides over Pd/POSS–TPAa catalysta
| Aryl halides |
Phenylboronic acid |
Products |
Time (h) |
Yieldb (%) |
| Reaction conditions: aryl halides (1 mmol), phenylboronic acid (1.5 mmol), Na2CO3 (0.15 g), deionized water (3 mL) and ethanol (3 mL), catalyst Pd/POSS–TPAa (0.0036 mmol), room temperature. The yield of product. |
 |
 |
 |
0.5 |
99 |
 |
 |
 |
0.5 |
86 |
 |
 |
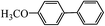 |
0.5 |
94 |
 |
 |
 |
4 |
85 |
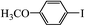 |
 |
 |
0.5 |
89 |
 |
 |
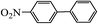 |
2 |
88 |
 |
 |
 |
2 |
88 |
 |
 |
 |
0.5 |
84 |
 |
 |
 |
2 |
86 |
 |
 |
 |
0.5 |
82 |
 |
 |
 |
0.5 |
80 |
 |
 |
 |
4 |
90 |
 |
 |
 |
4 |
93 |
 |
 |
 |
4 |
87 |
Catalyst reusability
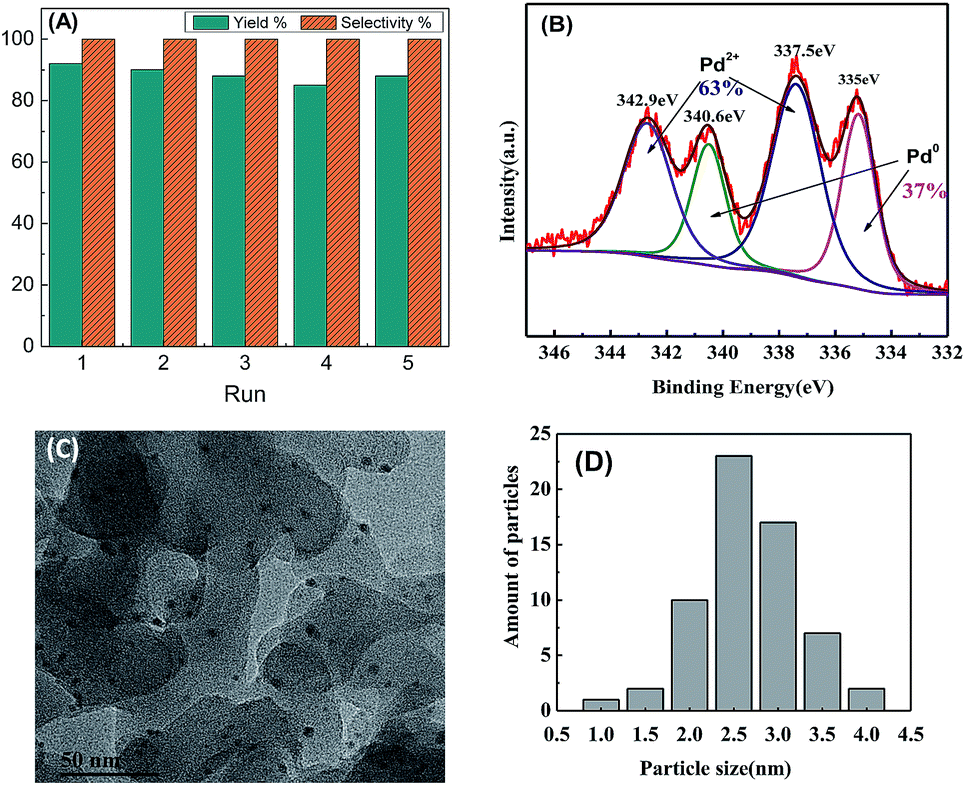
After the reaction, without any chemical or thermal activation, the catalyst can be reused after simple filtration and washing. The results of five-run recycling tests in Fig. 6A demonstrate a quite steady reusability of Pd/POSS–TPAa without observing significant loss in yield and selectivity. In order to verify the leaching of the Pd, a hot-filtration experiment was further carried out. After the Pd/POSS–TPAa catalyst was removed from the reaction solution after 5 min (yield: 51%), the supernatant did not show any further reactivity over the next 1 h. ICP-AES elemental analysis revealed 1.95 wt% Pd in the recovered catalyst, and the Pd species in the reaction media was less than 5 ppm, indicating the heterogeneous nature of the present catalyst. An XPS investigation on the recovered Pd/POSS–TPAa reveals four binding sites assignable to Pd2+ (342.9 and 337.5 eV) and Pd0 (340.6 and 335.0 eV) (Fig. 6B), which indicates that some Pd2+ species escape the strong coordination of amine/imines-to-Pd during catalysis and are subsequently converted to naked Pd0 due to the Pd2+/Pd0 catalytic cycle. The TEM image of the reused Pd/POSS–TPAa (Fig. 6C) shows that the Pd nanoparticles are highly dispersed throughout the polymer framework. The mean diameter of the particles was 2–3 nm (Fig. 6D), which was almost the same as that of the pristine catalyst. These features verify the strong coordination of Pd to the N-containing groups in cross-linked covalent network that may prevents the aggregation and leaching of the Pd and accounts for the steady reuse.
 |
| | Fig. 6 (A) Recycling runs for the coupling of bromobenzene with phenylboronic acid by Pd/POSS–TPAa. Reaction conditions were the same with that of Table 1; (B) XPS spectra of Pd 3d for reused Pd/POSS–TPAd; (C) TEM image and (D) particle size distribution of the recovered Pd/POSS–TPAa. | |
Conclusions
We have developed novel meso-/macroporous organic–inorganic porous covalent networks using Schiff base chemistry reaction. The resultant POSS–TPAx bear a number of attractive features, such as, high surface areas, large accessible porosities, and various N-containing functionalities, that exhibited an expected talent for supporting and stabilization of metal catalyst. The Pd trapped in POSS–TPAa led to the heterogeneous Suzuki–Miyaura reactions at room temperature with green features of outstanding catalytic activity, high TOF, convenient recovery, steady reuse, and mild reaction conditions. The strong coordination between Pd and the N-containing groups in covalent framework should be responsible for the excellent catalytic performance of Pd/POSS–TPAa. The new strategy this work may promote the development of both functional porous materials and advanced heterogeneous metal catalysts for multipurpose applications.
Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 21206052), MOE & SAFEA for the 111 Project (No. B13025), and the Fundamental Research Funds for the Central Universities (JUSRP51623A).
Notes and references
- F. Tang, L. Li and D. Chen, Adv. Mater., 2012, 24, 1504 CrossRef CAS PubMed.
- R. Li, A. Cao, Y. Zhang, G. Li, F. Jiang, S. Li, D. Chen, C. Wang, J. Ge and C. Shu, ACS Appl. Mater. Interfaces, 2014, 6, 20574 Search PubMed; Z. Li, D. Wu, Y. Liang, R. Fu and K. Matyjaszewski, J. Am. Chem. Soc., 2014, 136, 4805 CrossRef CAS PubMed.
- B. Dutta, S. Biswas, V. Sharma, N. O. Savage, S. P. Alpay and S. L. Suib, Angew. Chem., Int. Ed., 2016, 128, 1 CrossRef.
- W. Gao, Y. Chen, Y. Niu, K. Williams, L. Cash, P. J. Perez, L. Wojtas, J. Cai, Y. Chen and S. Ma, Angew. Chem., Int. Ed., 2014, 53, 2615 CrossRef CAS PubMed; B. Li, Y. Zhang, D. Ma, T. Ma, Z. Shi and S. Ma, J. Am. Chem. Soc., 2014, 136, 1202 CrossRef PubMed.
- W. Zhang, T. Liu, H. Wu, P. Wu and M. He, Chem. Commun., 2015, 51, 682 RSC; W. Mai, B. Sun, L. Chen, F. Xu, H. Liu, Y. Liang, R. Fu, D. Wu and K. Matyjaszewski, J. Am. Chem. Soc., 2015, 137, 13256 CrossRef CAS PubMed.
- M. Seino, W. Wang and J. E. Lofgreen, et al., J. Am. Chem. Soc., 2011, 133, 18082 CrossRef CAS PubMed; B. Karimi, M. Khorasani, H. Vali, C. Vargas and R. Luque, ACS Catal., 2015, 5, 4189 CrossRef.
- D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959 CrossRef CAS PubMed; S. Dutta, K. C. Wu and T. Kimura, Chem. Mater., 2015, 27, 6918 CrossRef; Q. Zhao, P. Zhang, M. Antonietti and J. Yuan, J. Am. Chem. Soc., 2012, 134, 11852 CrossRef PubMed; Q. Sun, Z. Dai, X. Meng, L. Wang and F. Xiao, ACS Catal., 2015, 5, 4556 CrossRef.
- D. Niu, Z. Liu, Y. Li, X. Luo, J. Zhang, J. Gong and J. Shi, Adv. Mater., 2014, 26, 4947 CrossRef CAS PubMed.
- J. Wang, W. Sng, G. Yi and Y. Zhang, Chem. Commun., 2015, 51, 12076 RSC; O. Buyukcakir, S. H. Je, D. S. Choi, S. N. Talapaneni, Y. Seo, Y. Jung, K. Polychronopoulou and A. Coskun, Chem. Commun., 2016, 52, 934 RSC.
- H. Lin, J. Ou, Z. Zhang, J. Dong and H. Zou, Chem. Commun., 2013, 49, 231 RSC.
- X. Wang, Y. Zhou, Z. Guo, G. Chen, J. Li, Y. Shi, Y. Liu and J. Wang, Chem. Sci., 2015, 6, 6916 RSC.
- G. Ding, W. Wang, T. Jiang and B. Han, Green Chem., 2013, 15, 3396 RSC; P. R. Boruah, A. A. Ali, B. Saikia and D. Sarma, Green Chem., 2015, 17, 1442 RSC; P. R. Boruah, A. A. Ali, M. Chetia, B. Saikia and D. Sarma, Chem. Commun., 2015, 51, 11489 RSC; W. Niu, Y. Gao, W. Zhang, N. Yan and X. Lu, Angew. Chem., 2015, 127, 8389 CrossRef CAS; Y. Wang, S. De and N. Yan, Chem. Commun., 2016, 52, 6210 RSC.
- L. Duan, R. Fu, Z. Xiao, Q. Zhao, J. Wang, S. Chen and Y. Wan, ACS Catal., 2015, 5, 575 CrossRef CAS; Z. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Mater., 2015, 27, 1921 CrossRef; X. Le, Z. Dong and Y. Liu, et al., J. Mater. Chem. A, 2014, 2, 19696 Search PubMed; Z. Jiao, Z. Zhai, X. Guo and X. Guo, J. Phys. Chem. C, 2015, 119, 3238 Search PubMed.
- L. Peng, J. Zhang, S. Yang, B. Han, X. Sang, C. Liu and G. Yang, Green Chem., 2015, 17, 4178 RSC; J. Chen, R. Liu, Y. Guo, L. Chen and H. Gao, ACS Catal., 2015, 5, 722 CrossRef CAS; P. Zhang, Z. Qiao, X. Jiang, G. M. Veith and S. Dai, Nano Lett., 2015, 15, 823 CrossRef PubMed.
- S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 17310 CrossRef CAS PubMed; P. Pachfule, M. K. Panda, S. Kandambeth, S. M. Shivaprasad, D. Diaz Diaz and R. Banerjee, J. Mater. Chem. A, 2014, 2, 7944 Search PubMed; A. Nagai, X. Chen, X. Feng, X. Ding, Z. Guo and D. Jiang, Angew. Chem., 2013, 125, 3858 CrossRef; X. Chen, N. Huang, J. Gao, H. Xu, F. Xu and D. Jiang, Chem. Commun., 2014, 50, 6161 RSC.
- S. Frindy, A. Primo, M. Lahcini, M. Bousmina, H. Garcia and A. El Kadib, Green Chem., 2015, 17, 1893 RSC; K. Mori, M. Dojo and H. Yamashita, ACS Catal., 2013, 3, 1114 CrossRef CAS.
- H. He, B. Li, J. Dong, Y. Lei, T. Wang, Q. Yu, Y. Feng and Y. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 8058 CAS.
- Q. Fang, S. Gu, J. Zheng, Z. Zhuang, S. Qiu and Y. Yan, Angew. Chem., Int. Ed., 2014, 53, 2878 CrossRef CAS PubMed; M. G. Schwab, B. Fassbender, H. W. Spiess, A. Thomas, X. Feng and K. Mullen, J. Am. Chem. Soc., 2009, 131, 7216 CrossRef PubMed.
- Z. Wang, J. Wei, G. Liu, Y. Zhou, K. Han and H. Ye, Catal. Sci. Technol., 2015, 5, 3926 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional reaction results and catalyst characterizations. See DOI: 10.1039/c6ra10641b |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.