DOI:
10.1039/C6RA08308K
(Paper)
RSC Adv., 2016,
6, 55786-55791
Cu1.5Mn1.5O4 spinel: a novel anode material for lithium-ion batteries†
Received
31st March 2016
, Accepted 27th May 2016
First published on 31st May 2016
Abstract
Copper manganese oxide Cu1.5Mn1.5O4, a novel anode material for lithium-ion batteries, was synthesized via a spray drying method. SEM images demonstrated a strong dependence of the morphologies of secondary and primary particles on the synthesis parameters. The surface electronic states and chemical compositions of manganese and copper in the sample were confirmed by XPS. The electrochemical results demonstrated that the sample synthesized at an annealing temperature of 700 °C showed great cycling performance, and retained a specific capacity of 464 mA h g−1 at a current of 100 mA g−1 after 60 cycles.
Introduction
In the last few decades, rapid growth in demand for lithium ion batteries (LIBs) for portable computers, hybrid electric vehicles (HEVs), pure electric vehicles (EVs) and plug-in hybrid vehicles (PHEVs) has required the drastic enhancement of their capacity and energy density.1–5 The theoretical capacity of a typical anode, graphite, is 372 mA h g−1 (for LiC6) and the practical capacity is 300–320 mA h g−1. Thus, there is a great demand for an alternative anode material that can exhibit a high reversible capacity.5
Much effort has been committed to the investigation of high capacity anode materials for LIBs. Among the candidates, metal oxides have attracted a lot of attention due to their advantageously high capacities. Though binary oxides have high capacities, there is also a severe volume expansion. Recently, a variety of ternary transition metal anode materials with a structural formula of AB2O4 have been widely investigated due to their electrochemical activity and stable framework.6 Studies have been reported on the compounds with the formula MCo2O4 (M = Ni,7 Mn,8 Fe,9 Cu10 or Mg11), MFe2O4 (M = Co,12 Ni,13 or Cu14), CoMn2O4, VFe2O4, etc. Like the ternary transition metal oxides, Cu1.5Mn1.5O4 also has a high theoretical capacity. Furthermore, copper and manganese are abundant, environmentally friendly, and relatively inexpensive compared to cobalt and nickel. Herein, Cu1.5Mn1.5O4 crystallizing with a spinel structure, Cu+[Cu0.52+Mn1.54+]O4, where Cu occupies the tetrahedral sites and Mn resides in the octahedral interstitial sites of the close-packed O2− ions,15,16 was investigated in this paper. To the best of our knowledge, Cu1.5Mn1.5O4 has only been used as a catalyst, as referred to in previous reports.17–19 So far, few studies have focused on this material, and especially its electrochemical performance, when applied in LIBs.
In our work, we report the use of spray drying methods for preparing Cu1.5Mn1.5O4. The objective of this work is to investigate the charge/discharge mechanism of Cu1.5Mn1.5O4. Also, the evaluation of the electrochemical performance of lithium cells assembled with this material as working electrodes will be discussed.
Experimental
Spray drying method
The compound Cu1.5Mn1.5O4 was prepared by a typical spray drying (SD) method using a citrate ligand to complex the metal ions. For this purpose, Cu(Ac)2·H2O (0.05 mol), Mn(Ac)2·4H2O (0.1 mol) and citric acid (0.05 mol) were dissolved in 125 mL of deionized water with vigorous agitation. The mixed solution was spray-dried as the outlet temperature was set to 185 °C. Then, the obtained green acetate powder was pre-sintered at 300 °C in air for 3 hours to decompose the organic constituents. After being ground to form powders, the precursor was sintered at different temperatures (700, 860, and 900 °C) in air for 21 hours to obtain the desired integrated crystalline samples (marked SD700, SD860, and SD900, respectively). The model of the spray drying equipment was SP-1500, Shunyi Shanghai Experimental Equipment Co. Limited.
Characterization
The phase composition and structure of the samples prepared at different temperatures were investigated using an X-ray diffractometer equipped with Cu Kα radiation (XRD, Rint 1000, Rigaku, Japan) over the 2θ range of 10–80°. The morphologies and sizes of the samples were directly examined by scanning electron microscopy (SEM). The tap density of samples was tested by a tap density tester (QL/BT303, Yongda). The size distribution of the particles of samples was tested by a laser particle size analyzer (Malvern, MASTERSIZER 3000). X-ray photoelectron spectroscopy (XPS, ESCA-LAB 250Xi) was used to confirm the valence states of Mn and Cu in the sample.
A CR2032 coin-tape cell was used to investigate the electrochemical performances of the synthesized materials. The active material was mixed in slurry containing 10 wt% Super P as a conductive agent and 10 wt% polyvinylidene fluoride (PVDF) as a binder. The homogeneous slurries were cast onto copper foils to obtain an electrode laminate which was dried at 110 °C in a vacuum drying oven overnight. The mass of the active material in the working electrode is about 9.48 mg, and the geometrical area of the electrode is 176.6 mm2. The working cell was assembled in a glove box filled with dry argon. A lithium disk (Φ 15 × 1 mm) was used as a negative electrode (counter electrode and reference electrode). A Celgard 2400 porous polypropylene film served as a separator. The electrolyte was 1 M LiPF6 dissolved in a compound of diethyl carbonate/ethylene carbonate (DEC/EC, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume). Galvanostatic charge–discharge cycling and cyclic voltammetry were tested at room temperature (RT ∼ 25 °C) by means of a computer-controlled battery evaluation system (LAND CT 2001, Wuhan, China).
:1 by volume). Galvanostatic charge–discharge cycling and cyclic voltammetry were tested at room temperature (RT ∼ 25 °C) by means of a computer-controlled battery evaluation system (LAND CT 2001, Wuhan, China).
Result and discussion
The samples synthesized by the spray drying method at different temperatures were analyzed by XRD, and the results are shown in Fig. 1. Narrow reflection peaks indicate high crystallinity for all three samples and the main peaks can be indexed to cubic Cu1.5Mn1.5O4 (JCPDS 35-1171, space group: Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m 227). Moreover, some weak peaks marked with star are observed in the XRD pattern of SD700, which can be ascribed to the existence of Mn2O3;17,19 while the impurity of Mn3O4 appears in the samples of SD860 and SD900, as indicated in their XRD patterns, which can be attributed to oxidation loss caused by the elevated annealing temperature.
m 227). Moreover, some weak peaks marked with star are observed in the XRD pattern of SD700, which can be ascribed to the existence of Mn2O3;17,19 while the impurity of Mn3O4 appears in the samples of SD860 and SD900, as indicated in their XRD patterns, which can be attributed to oxidation loss caused by the elevated annealing temperature.
 |
| | Fig. 1 XRD patterns of samples synthesized at different temperatures (* indicates lines due to Mn2O3 and • indicates lines due to Mn3O4). | |
The morphologies of the samples annealed at different temperatures were investigated and the corresponding SEM images are presented in Fig. 2a–c. The bulk particles in the SD700 are constituted by the aggregation of primary particles in the size range of 500–600 nm. As the annealing temperature increases, significant growth of particles is observed, as revealed in the SEM image for SD860. When the annealing temperature reached 900 °C, the apparent agglomeration of particles was detected so that the size of particles exceeded 3 μm. With the elevation in annealing temperature, the size of bulk particles constituted by the aggregation of 500–600 nm primary oxide particles increase distinctly, which demonstrates that the annealing temperature should have a significant influence on the morphologies and size of the oxide. We speculate that the sample annealed at 700 °C with the smallest particle size has the best electrochemical performance. The tap densities of SD700, SD860 and SD900 are 1.56, 1.84 and 2.10 g cm−3, respectively, which are higher than nano-size silicon, indicating that the material has a high volume energy density. The size distribution of the particles of the three samples were tested and the data are shown in Table 1. The data reveals that all three samples’ size distributions are larger than the results obtained from SEM, due to the aggregation of primary particles. The sample SD700 has the smallest size distribution, while the sample SD900 has the largest size distribution, which agrees well with the results of SEM.
 |
| | Fig. 2 SEM images of SD700 (a), SD860 (b) and SD900 (c). | |
Table 1 The size distribution of the particles of the three samples annealed at different temperatures
| |
Dv10 (μm) |
Dv50 (μm) |
Dv90 (μm) |
| SD700 |
1.17 |
7.25 |
39.6 |
| SD860 |
5.77 |
45.4 |
105.3 |
| SD900 |
10.6 |
46.3 |
121 |
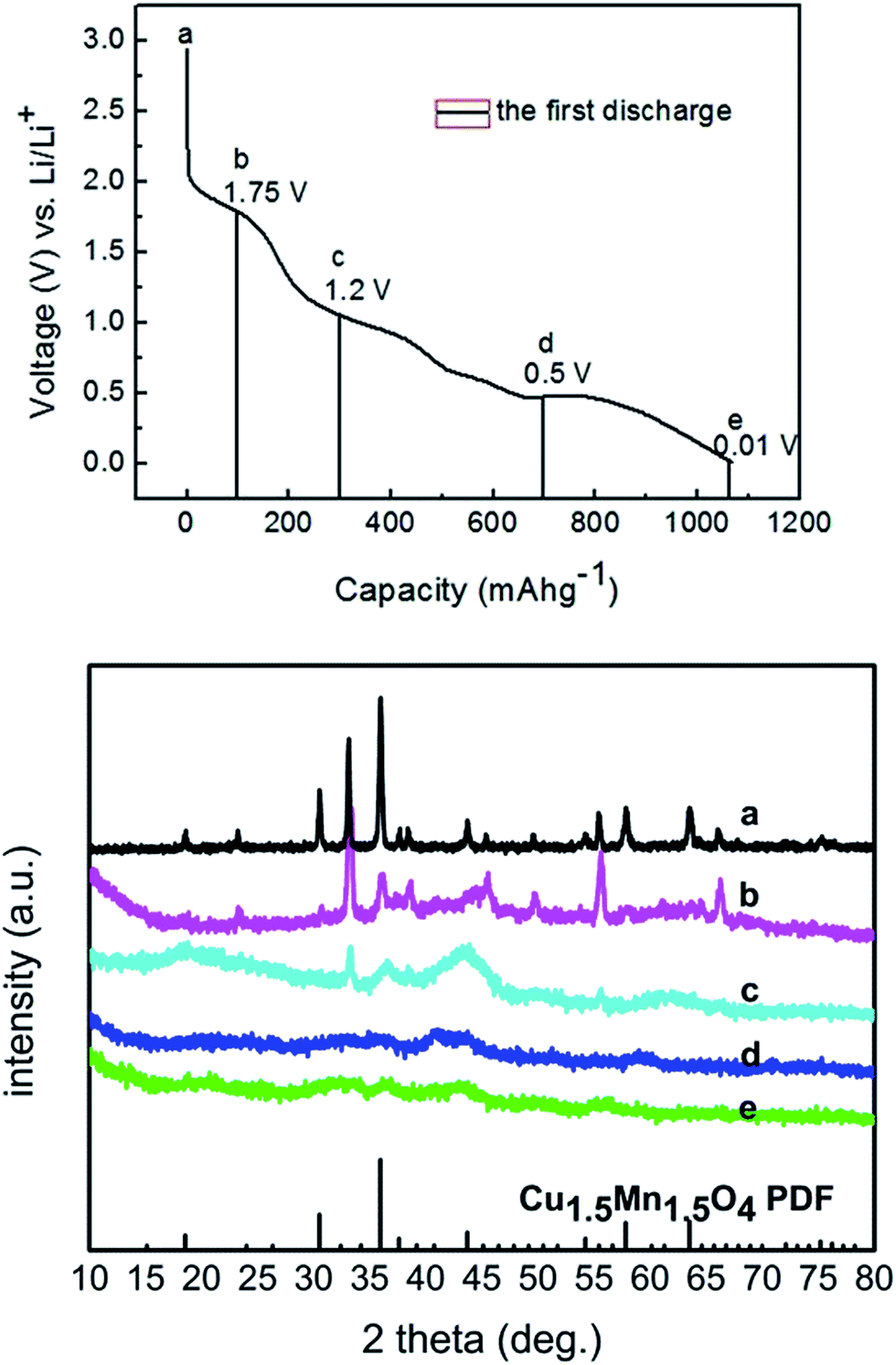
Ex situ XRD analysis was conducted on the electrodes after discharge/charge to selected potentials, as demonstrated in Fig. 3 and 4. The diffraction peaks referring to Cu1.5Mn1.5O4 broadened gradually and finally disappeared during the discharge process, indicating that the crystal structure was destroyed, which was similar to the results concerning NiFe2O420 and CoFe2O421 reported in the literature. After being charged back to 0.5 V, only broad peaks assigned to an amorphous phase were preserved without any trace of Cu1.5Mn1.5O4 existing, further indicating the irreversible structural change during the first discharge process. Thus, it was demonstrated that the reaction mechanism was a redox reaction in which the structure of crystal Cu1.5Mn1.5O4 was destroyed and atomic-scaled mixing of metallic Cu and Mn particles along with Li2O occurred after the first discharge process, which proved to be irreversible.
 |
| | Fig. 3 XRD pattern of the SD700 sample during discharge process: (a) the SD700 electrode; (b) discharged to 1.75 V; (c) discharged to 1.2 V; (d) discharged to 0.5 V; (e) discharged to 0.01 V during the first cycle. | |
 |
| | Fig. 4 XRD pattern of the SD700 sample during charge process: (a) charged to 0.5 V; (b) charged to 1.25 V; (c) charged to 3 V during the first cycle. | |
The electrochemical performance of these samples was further investigated to verify the dependence on the particle size. The discharge/charge profiles of these three samples are presented in Fig. 5. Testing was operated in the potential range of 0.01–3 V at the current rate of 100 mA g−1. The first discharge curve shows an initial abrupt decrease of the cell potential followed by some plateaus, during which the exhibited overall capacity corresponds well to the electrons needed for the reduction of transition metals involved. As the discharge process proceeds, the potential decreases more steeply until it reaches the end value of 0.01 V. As shown, the first discharge capacities of the samples SD700, SD860, and SD900 are 1031.7, 1025.0, and 1019 mA h g−1 respectively, while the reversible charge capacities are 664.1 (about 64.4%), 582.6 (about 56.8%), and 574.4 mA h g−1 (about 56.4%), demonstrating irreversible capacity losses of 367.6 mA h g−1 (about 35.6%), 442.4 mA h g−1 (about 43.2%), and 444.7 mA h g−1 (about 43.6%), respectively. The high reversible capacity of the SD700 sample, that was comprised of small particles, can be attributed to a much larger surface area and a much shorter Li-ion insertion length, which evokes a great enhancement in its coulombic efficiency. That is to say, the well-adjusted sintering temperature for synthesis is crucial in determining the corresponding electrode performance of the as-prepared sample. The large irreversible capacity loss in the first cycle can be attributed to the formation of the SEI film onto the surface of the oxide electrode22 and the lack of further oxidation of Mn2+ to Mn3+ and Mn4+. The reversible capacity of SD700 in the subsequent cycles is 664.1 mA h g−1, which is close to the theoretical capacity of Cu1.5Mn1.5O4 (665.1 mA h g−1, according to eqn (1) and (3)) and Mn2O3 (678.6 mA h g−1, according to eqn (2) and (4)).
| | |
Cu1.5Mn1.5O4 + 8Li → 1.5Cu + 1.5Mn + 4Li2O
| (1) |
| | |
Mn2O3 + 6Li → 2Mn + 3Li2O
| (2) |
| | |
1.5Cu + 1.5Mn + 3Li2O → 1.5CuO + 1.5MnO + 6Li
| (3) |
| | |
2Mn + 2Li2O → 2MnO + 4Li
| (4) |
 |
| | Fig. 5 (a), (b) and (c) The discharge/charge profiles of SD700, SD860, and SD900, respectively. (d) Cycling performance of the samples obtained at different temperatures in the voltage range of 0.01–3.0 V vs. Li/Li+ at current densities of 100 mA g−1. | |
The cycling performance of the samples synthesized at different temperatures is shown in Fig. 5d. A fast capacity fading in the first 25 cycles is observed, possibly due to complicated side-reactions and/or irreversible structure transformation. Afterwards, the capacity is stabilized because the electrochemically reversible structures for the Li-ion insertion reaction have been established. Those discharge capacities are 464, 406, 404 mA h g−1 for the samples SD700, SD860 and SD900 after 60 cycles, respectively, which are all higher than the capacity provided by graphite (about 372 mA h g−1). The sample of SD700, with the smallest particle size, has the best capacity retention, which can be attributed to the appropriate particle sizes, inducing the increased electrochemically active surface area, and the structural strains in accommodating Li+.23 The SD700 sample was used in the following characterizations for the clear observation and illustration of the reaction mechanism, due to its good electrochemical performance.
An XPS study was carried out to confirm the surface electronic states and chemical compositions of manganese and copper in the sample synthesized at an annealing temperature of 700 °C. Fig. 6 shows the Mn 2p spectrum and Cu 2p spectrum. As shown in Fig. 6a, the binding energy of the main Mn 2p3/2 line appears at 641.7 eV, an intermediate between that characteristic of Mn3+ species and that characteristic of Mn4+ species.24 This can be deconvoluted into two peaks at 641.1 eV and 643.1 eV, which can be indexed to Mn3+ 2p3/2 and Mn4+ 2p3/2, respectively, while a peak matching to Mn2+ 2p3/2 is not detected. Fig. 6b shows the Cu 2p spectrum for the sample SD700, which can be deconvoluted into a relatively narrow peak at 930.8 eV and a broader peak at 933.8 eV. An evident shake-up satellite structure is observed at 940.7 eV and 943.7 eV. The broader peak and the satellite structure are assigned to Cu2+ 2p3/2, while the narrow peak at a lower binding energy indicates the presence of Cu+ 2p3/2,24,25 which can be evidence for the successful fabrication of Cu1.5Mn1.5O4 in our experiment.
 |
| | Fig. 6 (a) Mn 2p spectrum recorded from the sample SD700. (b) Cu 2p spectrum recorded from the sample SD700. | |
Cyclic voltammetry measurements were conducted to observe the redox process during the first 5 cycles, as depicted in Fig. 7. In the first cathodic cycle, a broad peak centered at 1.3 V is observed, which can be attributed to the reduction of Mn4+ and Mn3+ to Mn2+.14 In addition, the broad peak at 0.75 V is attributed to the reduction of Cu2+ and Cu+ to Cu,26 while the minor peak at 0.4 V corresponds to the formation of the solid electrolyte interface (SEI). An intense peak emerges as it cycles to 0.25 V, which is ascribed to the reduction of Mn2+ to metallic Mn embedded in the Li2O matrix.14 There are two oxidation peaks in the anodic sweep: a broad one at 2.0 V corresponding to the oxidation of Cu to Cu2+,26 and the other one at 1.3 V due to the oxidation of Mn to Mn2+.14 However, the current responses for these redox couples decrease significantly in the following cycles, demonstrating that an irreversible phase transformation occurs in the initial cycle. Besides, the intensities of anodic/cathodic peaks associated with the Mn2+/Mn at 0.32/1.35 V and Cu2+/Cu at 0.95/1.82 V remain almost constant, indicating the good reversibility of the electrochemical reactions in subsequent cycles. The current response for Mn4+ or Mn3+ to Mn2+ reduction cannot be observed, which is consistent with the fact that the Mn in the lower oxidation state cannot be oxidized to a state over +2 in the potential range of 0.01–3.0 V.27
 |
| | Fig. 7 The first five consecutive CV scans of the sample SD700 electrode (in the voltage range of 0.01–3.0 V vs. Li/Li+ at a scan rate of 0.1 mV s−1). | |
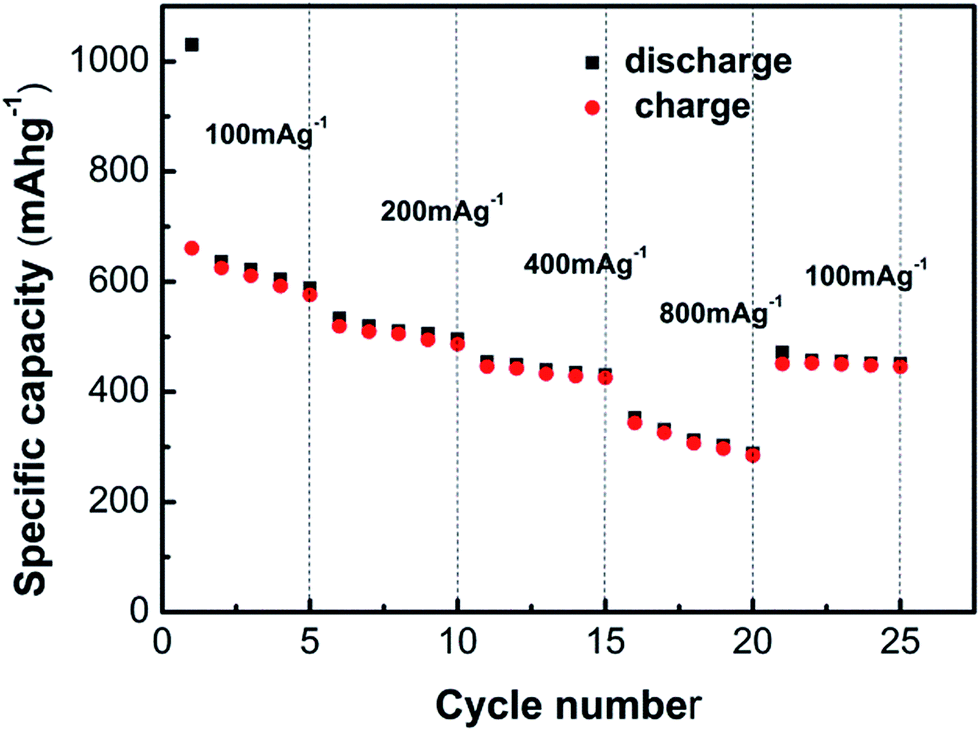
The rate capability of the anode material SD700 was investigated, as shown in Fig. 8, which was carried out at varied current rates of 100, 200, 400 and 800 mA h g−1 subsequently. The observed capacities of SD700 are 613.7 mA h g−1 at 100 mA g−1, 503.28 mA h g−1 at 200 mA g−1, 435.2 mA h g−1 at 400 mA g−1, and 311.6 mA h g−1 at 800 mA g−1, respectively. The exhibited reversible capacity and rate capacity of Cu1.5Mn1.5O4 is acceptable in comparison with other ternary transition metal anode materials. Nevertheless, it should be pointed out that an improvement in coulombic efficiency for the first cycle is necessary, possibly through surface modification, and further efforts towards this are still in progress.
 |
| | Fig. 8 Rate capacity of the sample SD700 at various current densities from 100 mA g−1. | |
Conclusions
In summary, a facile spray drying method and annealing method has been developed for the synthesis of Cu1.5Mn1.5O4. Cu1.5Mn1.5O4 has been reported for the first time as an anode material for LIBs, and exhibits good electrochemical performance with a high specific charge capacity of 464 mA h g−1 after 60 cycles at the current rate of 100 mA h g−1. The involved reaction mechanism of Cu1.5Mn1.5O4 has been investigated and demonstrated as the conversion reaction mechanism by ex situ XRD, CV and XPS analysis. In addition, the calcination temperature of the as-prepared samples has been explored to optimize the electrochemical performance. The as-prepared SD700 displays a good performance and is considered to be a promising anode material for lithium ion batteries.
Acknowledgements
This work was financially supported by the Ministry of Science and Technology of People’s Republic of China (contract number: 2010EG111015) and Science & Technology Office of Jiangsu Province (BE2013006-3).
References
- T. Ohzuku and A. Ueda, J. Electrochem. Soc., 1994, 141, 2972 CrossRef CAS.
- X. Li and C. Wang, J. Mater. Chem. A, 2013, 1, 165–182 CAS.
- Y. S. Hu, P. Adelhelm, B. M. Smarsly, S. Hore, M. Antonietti and J. Maier, Adv. Funct. Mater., 2007, 17, 1873 CrossRef CAS.
- Y. Zhao, X. Li, B. Yan, D. Xiong, D. Li, S. Lawes and X. Sun, Adv. Energy Mater., 2016, 6 DOI:10.1002/aenm.201502175.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364 CrossRef CAS PubMed.
- X. Li and C. Wang, RSC Adv., 2012, 2, 6150–6154 RSC.
- T. Yang, D. Xia, Z. Wang and Y. Chen, Mater. Lett., 2009, 63, 5 CrossRef CAS.
- P. Lavela, J. L. Tirado and C. Vidal-ABarca, Electrochim. Acta, 2007, 52, 7986 CrossRef CAS.
- Y. Sharma, N. Sharma, G. V. Subba Rao and B. V. R. Chowdari, Solid State Ionics, 2008, 179, 587 CrossRef CAS.
- Y. Sharma, N. Sharma, G. V. Subba Rao and B. V. R. Chowdari, J. Power Sources, 2007, 173, 495 CrossRef CAS.
- P. Lavela, J. L. Tirado, M. Womes and J. C. Jumas, J. Electrochem. Soc., 2009, 156, A589 CrossRef CAS.
- P. Lavela and J. L. Tirado, J. Power Sources, 2007, 172, 379 CrossRef CAS.
- M. Bomio, P. Lavela and J. L. Tirado, J. Solid State Electrochem., 2008, 12, 729 CrossRef CAS.
- L. Zhou, D. Zhao and X. W. Lou, Adv. Mater., 2012, 24, 745 CrossRef CAS PubMed.
- V. Koleva, D. Stoilova and D. Mehandjiev, J. Solid State Chem., 1997, 133, 416 CrossRef CAS.
- S. Vepřek, D. L. Cocke, S. Kehl and H. R. Oswald, J. Catal., 1986, 100, 250 CrossRef.
- R. He, Q. Liu, K. Zhi and X. Cui, J. Fuel Chem. Technol., 2008, 36, 767 CAS.
- I. Spassova and D. Mehandjiev, React. Kinet. Catal. Lett., 2000, 69, 223 CrossRef.
- N. M. Deraz and O. H. Abd-Elkader, Int. J. Electrochem. Sci., 2013, 8, 10112 CAS.
- Y. N. NuLi and Q. Z. Qin, J. Power Sources, 2005, 142(1–2), 292 CrossRef CAS.
- M. Bomio, P. Lavela and J. Tirado, ChemPhysChem, 2007, 8, 1999 CrossRef CAS PubMed.
- R. Dedryere, S. Laruelle, S. Grugeon, P. Poizot, D. Gonbeau and J.-M. Tarascon, Chem. Mater., 2004, 16, 1056 CrossRef.
- Y. Ding, Y. Yang and H. Shao, Solid State Ionics, 2012, 217, 27 CrossRef CAS.
- J. L. Gautier, E. Rios, M. Gracia, J. F. Marco and J. R. Gancedo, Thin Solid Films, 1997, 311, 51 CrossRef CAS.
- P. A. Wright, S. Natarajan, M. Thomas and P. L. Gai-Boyes, Chem. Mater., 1992, 4, 1053 CrossRef CAS.
- M. V. Reddy, C. Yu, F. Jiahuan, K. Ping Loh and B. V. R. Chowdari, RSC Adv., 2012, 2, 9619 RSC.
- F. M. Courtel, Y. Abu-Lebdeh and I. J. Davidson, Electrochim. Acta, 2012, 71, 123 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra08308k |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.