
Non-enzymatic glycation mediated structure–function changes in proteins: case of serum albumin
Abstract
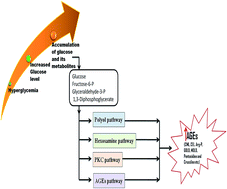
Albumin, a major plasma protein with extraordinary ligand binding properties, transports various ligands ranging from drugs, hormones, fatty acids, and toxins to different tissues and organs in the body. Albumin can undergo glycation on reacting with circulating reducing sugars, and this modification can occur even more frequently under hyperglycaemic conditions. Glycation is a non-enzymatic reaction of proteins and sugars, which gives rise to the formation of early and advanced glycation end products (AGEs). AGEs are a fundamental factor in the development and progression of severe diabetic complications such as nephropathy, neuropathy, retinopathy and cardiovascular disorders. The pathological impact of AGEs is mainly attributed to the structural modifications of proteins due to their cross-linking properties and the modification of lysine and arginine side chains. Herein, we review the structure–function effects of glycation on albumin, specifically focusing on its drug binding functions. Considering its physiological significance, there is a need to focus current research on determining the structure–function impacts of non-enzymatic glycation on serum albumin.
 Please wait while we load your content...
Please wait while we load your content...

