
Selective topotecan delivery to cancer cells by targeted pH-sensitive mesoporous silica nanoparticles†
Abstract
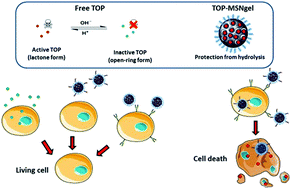
Topotecan (TOP), a water-soluble derivative of camptothecin, is a potent antitumor agent that is receiving growing attention for the treatment of several types of cancer. However, one of the major constraints in the clinical use of this drug is its inactivation at the physiological pH of 7.4. Mesoporous silica nanoparticles (MSNs) constitute promising nanocarriers to circumvent this issue. Herein TOP has been encapsulated into MSNs and the nanosystem has been provided with selectivity towards tumor cells, which permits releasing the active form of the molecule at the acidic cell compartments (endo/lysosomes; pH ≤ 5.5) following nanoparticle internalization. For this purpose, MSNs have been coated with a multifunctional gelatin shell that: (i) protects TOP from hydrolysis and prevents its premature release; (ii) acts as a pH-sensitive layer; and (iii) provides multiple anchoring points for the grafting of targeting ligands, such as folic acid (FA), for selective internalization in tumor cells. In vitro tests demonstrate that cancer cells that overexpress membrane cell surface markers with affinity towards FA, internalize a higher percentage of nanoparticles than healthy cells, which do not overexpress such markers. Moreover, the nanosystems are efficient at killing tumor cells, whereas they do not decrease the viability of normal cells. In contrast, free TOP failed to kill both cell lines, which can be ascribed to the inactivation of the drug. This novel nanodevice constitutes a step forward toward the design of novel weapons to fight against cancer.
 Please wait while we load your content...
Please wait while we load your content...

