
Synthesis of copper–silver bimetallic nanopowders for a biomedical approach; study of their antibacterial properties
Abstract
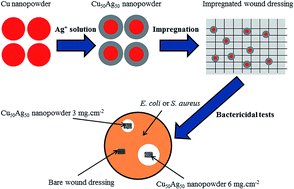
Copper–silver nanopowders (NPs) are synthesized using a combination of sonoelectrodeposition for the inner core and galvanic replacement reaction for the outer shell. These combined techniques provide pure copper core NPs and allow the replacement of the surface copper atoms by silver atoms. The different Cu–Ag NPs are characterized by transmission electron microscopy, centrifugal liquid sedimentation, X-ray diffraction, X-ray photoelectron spectroscopy and energy dispersive X-ray spectroscopy. The diameter of NPs is approximately equal to 7 nm. X-ray photoelectron spectroscopy results tend to support a core–shell structure. The bactericidal properties of these NPs are tested against both Staphylococcus aureus (Gram-positive) and Escherichia coli (Gram-negative) bacteria. The efficiency of these NPs seems essentially due to the release of Cu2+ and Ag+ ions. The study of different compositions of Cu–Ag NPs exhibits a good compromise against both S. aureus and E. coli when the silver atomic percentage is superior to 40%. Finally, tests of wound dressings impregnated with Cu50Ag50 NP are performed and exhibit successfully their bactericidal properties.
 Please wait while we load your content...
Please wait while we load your content...

