
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoformulations of anticancer thiosemicarbazones to reduce methemoglobin formation and improve anticancer activity†
Britta
Fischer‡
a,
Kushtrim
Kryeziu‡
b,
Sebastian
Kallus
a,
Petra
Heffeter
*bc,
Walter
Berger
bc,
Christian R.
Kowol
*ac and
Bernhard K.
Keppler
ac
aInstitute of Inorganic Chemistry, University of Vienna, Waehringer Str. 42, 1090 Vienna, Austria. E-mail: christian.kowol@univie.ac.at; Fax: +43-1-4277-52680; Tel: +43-1-4277-52609
bInstitute of Cancer Research and Comprehensive Cancer Center, Medical University Vienna, Borschkegasse 8a, 1090 Vienna, Austria. E-mail: petra.heffeter@meduniwien.ac.at; Fax: +43-1-40160-957555; Tel: +43-1-40160-57557
cResearch Platform “Translational Cancer Therapy Research”, University of Vienna, Medical University of Vienna, Vienna, Austria
First published on 2nd June 2016
Abstract
Triapine is a promising anticancer thiosemicarbazone which was investigated in multiple clinical trials, where it was active against leukemia but not against solid cancers. This is probably based on insufficient drug levels in the tumor due to a short plasma half-life. Therefore, we encapsulated Triapine into polymeric nanoparticles and remote-loaded liposomes to improve the drug pharmacokinetics as well as targeted delivery. However, burst release of Triapine from both nanoformulations was observed, making the synthesis of two novel Triapine derivatives necessary in order to improve the remote-loading properties. Indeed, the encapsulation efficiency increased and for one derivative also the desired continuous drug release was in line with a strongly reduced cytotoxic activity against cancer cells. In vivo studies of this most promising formulation demonstrated a significant increase in survival of the animals compared to the free drug. Finally, we investigated drug-induced methemoglobin formation, a frequently observed side effect of thiosemicarbazones, which was completely prevented by the liposomal formulation.
Introduction
α-N-heterocyclic thiosemicarbazones are promising candidates for cancer therapy. Regarding the mode of action, it is assumed that they interact with (intracellular) iron via their tridentate NNS donor atoms. One enzyme, which is known to be very sensitive to disturbance of the iron levels, is ribonucleotide reductase (RR), responsible for the synthesis of DNA building blocks.1 Since fast proliferating tumor cells are frequently characterized by RR overexpression, this enzyme represents a promising and convenient target for cancer chemotherapy.2 It is believed that intracellularly formed thiosemicarbazone–iron complexes destroy the tyrosyl radical of the R2 subunit of RR (probably by direct interaction).3 However, treatment of cancer cells with thiosemicarbazones also resulted in several other effects like cell cycle arrest via c-myc and p21 as well as activation of diverse stress kinase pathways.4 Finally, some thiosemicarbazones have also been suggested to interact with the cellular copper homeostasis.5,6With regard to clinical investigations, 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (Triapine; Scheme 1) is the most prominent representative of α-N-heterocyclic thiosemicarbazones, as it has been already investigated in more than 30 clinical phase I/II trials. In addition, currently a novel thiosemicarbazone, di-2-pyridylketone-4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC) is entering clinical phase I studies.7 However, so far no iron chelation-based therapy has been approved for cancer therapy. In case of Triapine, it is also worth noting that the clinical evaluation revealed mainly activity against hematological cancer types (e.g. advanced leukemia8,9), but showed disappointing results as monotherapy against solid tumor types like non-small-cell lung cancer10 and renal cell carcinoma.11 The exact reasons are still unknown. We were recently able to show that Triapine is characterized by a very short plasma half-life time, which together with its transient activity results in rapid tumor re-growth in mice upon therapy cessation.12 Thus, the current hypothesis is that plasma levels of Triapine are probably insufficient for drug delivery and long-lasting maintenance of RR inhibition in solid tumor nodules. In addition, clinical trials indicated that interaction of Triapine with the iron-rich red blood cell compartment leads to the development of methemoglobinemia in the majority of the treated patients.13 Consequently, there is an urgent need for new strategies to increase the plasma half-life and tumor-specificity of thiosemicarbazones.
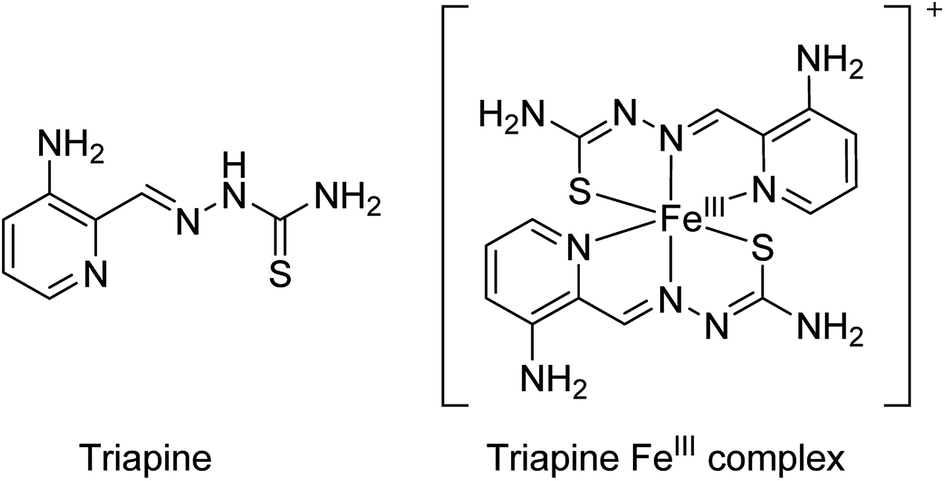 | ||
| Scheme 1 Triapine and its FeIII complex. | ||
Nanoparticulate drug formulations would be an ideal strategy, as encapsulation of the drug would offer distinctly prolonged plasma half-life, and additionally specific tumor accumulation via the enhanced permeability and retention (EPR) effect.14 This effect allows nanoformulations to enrich in the tumor tissue due to leaky blood vessels and the absence of an effective lymphatic drainage system.15
In this study, we investigated, on the one hand, encapsulation of Triapine into the polymer matrices poly(lactic acid) (PLA) and poly(lactide-co-glycolide) (PLGA), since, both are biodegradable, biocompatible and are approved by the US Food and Drug Administration (FDA) as therapeutic drug carriers in humans.16 On the other hand, we tested encapsulation into liposomal formulations by the remote loading approach, which is one of the best-studied preparation methods for liposomes with e.g. Doxil® (liposomal doxorubicin) as clinically approved representative.17 In addition, two novel Triapine derivatives were synthesized possessing a weak base amine, as such modifications were already reported to improve the loading and stability of liposomal formulations in case of gemcitabine18 or docetaxel.19 The novel nanoformulations were physico-chemically characterized and the drug-release behavior was correlated with the cytotoxicity in comparison to the free drugs. For the most promising formulation, cell uptake studies were performed and the anticancer activity was evaluated in vivo. Finally, the ability of the nanoformulation to prevent methemoglobin formation was studied.
Results and discussion
Polymeric nanoparticles – preparation and characterization
Two different Triapine-loaded nanoparticles were synthesized using the nanoprecipitation method. As polymer matrices, the biocompatible and biodegradable polylactic acid (PLA) and poly(lactide-co-glycolide) (PLGA) were used. The encapsulation of Triapine was performed by addition of acetone solutions of the respective polymer and the drug to an aqueous solution of the surfactant (Tween 80). The nanoparticles were produced in triplicate and characterized concerning their size, polydispersity index (PDI), zeta potential and encapsulation efficiency (Table S1†). The dynamic light scattering (DLS) data showed particle sizes between 110 and 130 nm (ESI Fig. S1 and S2†) with a PDI < 0.15 indicating very homogeneous nanoformulations. The electrostatic repulsion of the particle surfaces (zeta potential) was negative at −14 ± 1 mV for the PLA and −8 ± 1 mV for the PLGA nanoparticles, which is in accordance with the presence of terminal carboxylic acid groups in the polymers.20 The encapsulation efficiency (EE) of Triapine was determined right after production by evaporation of the solvent and dissolving the thin film in a mixture of acetone and dichloromethane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The resulting solution was analyzed for Triapine content by UV-Vis spectroscopy revealing a quite low EE for both polymers (<25%).
:1). The resulting solution was analyzed for Triapine content by UV-Vis spectroscopy revealing a quite low EE for both polymers (<25%).
Drug release and anticancer activity of Triapine-loaded nanoparticles
Drug release from the Triapine nanoparticles was investigated using the dialysis bag diffusion technique.21 To this end, the concentration of released Triapine in the outer compartment was measured by fluorescence spectroscopy at various time points with an excitation wavelength of 368 nm and an emission wavelength of 455 nm. Noteworthy, the gained release data from these particles is strongly influenced, on the one hand, by the partition coefficient of the drug between the polymer and the aqueous phase and, on the other hand, by diffusion flux of the drug across the membrane.22 Consequently, dialysis of free Triapine was performed as a reference and accounted for 100% drug release. After two hours under physiological conditions at 37 °C, ∼80% of Triapine was released from PLGA and 96% from PLA nanoparticles (Fig. S3†). This indicates that there are either only weak interactions between the drug and the polymer23 and/or a high surface location of the drug.24In line with these data, MTT assays performed on SW480 colon and A2780 ovarian cancer cells after 72 h drug incubation revealed no difference in the activity of Triapine-loaded nanoparticles to free Triapine (Table S2 and Fig. S4†). As expected, the unloaded nanoparticles did not show any activity in these tests. Notably, amphiphilic co-polymer nanoparticles with 2-benzoylpyridine 4-ethyl-3-thiosemicarbazone also showed similar cytotoxicity compared to the free drug after 48 h incubation,25 although after 48 h only ∼50% of the drug was released in PBS pH 7.4.
Overall, these data indicate that, although, drug encapsulation of Triapine within polymeric nanoparticles has been successfully achieved, the resulting nanoparticles showed insufficient stability for biological use. Hence, encapsulation into liposomes as an alternative strategy of nanoformulation was investigated.
Liposomal formulations
One of the best studied preparation methods for liposomes is the so-called remote loading approach, which is also for example used in the preparation of Doxil® (encapsulated doxorubicin) as the most prominent clinically approved representative.17 In general, doxorubicin can be considered an ideal drug for this loading approach, since it contains a crucial weak base amine that is protonated inside the ammonium sulfate-containing (∼pH 5) liposomes. Moreover, in order to ensure optimal remote loading by the ammonium gradient, the loaded molecules should have a logD at pH 7 in the range of −2.5 to 2 and a pKa ≤ 11.26 Also Triapine fits to these prerequisites with a logD7.4 = 0.85 ± 0.08 (ref. 27) and a pKa ≤ 11 (pK1 = 3.92 and pK2 = 10.78).28 However, as the pyridine pK1 = 3.92 is quite low, we hypothesized that Triapine might only be partially protonated inside the liposomal core. Hence, two additional novel Triapine derivatives were synthesized (Scheme 2) containing a short lipophilic tail as well as a weak base amine to introduce an additional protonatable moiety in order to achieve properties more comparable to doxorubicin.
 | ||
| Scheme 2 Synthesis of Triapine derivatives 1 and 2. | ||
Synthesis and characterization of Triapine derivatives
The Triapine derivatives were synthesized in multiple steps as depicted in Scheme 2. The first step for the preparation of 2 included tert-butyloxycarbonyl (BOC) protection of one NH2 group (certainly not necessary in case of 1). Further on, both compounds were prepared according to the same strategy. The respective thiosemicarbazides were obtained by reaction of the amines with CS2, followed by treatment with CH3I and displacement of the S-methyl group by hydrazine hydrate at 90 °C in ethanol. Subsequent condensation with the protected 3-amino-2-pyridine carboxaldehyde and acidic deprotection, led to the desired thiosemicarbazones 1 and 2. The compounds were characterized by 1H-NMR, 13C-NMR, as well as elemental analysis (see Experimental section).Liposome preparation and characterization
The lipid composition of the liposomes was chosen in accordance to Doxil17 using DSPC/CHOL/mPEG2000-DSPE in molar ratios of 56:39:5 (DSPC = 1,2-distearoyl-sn-glycero-3-phosphocholine; CHOL = cholesterol; DSPE-mPEG(2000) = 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000]). Prior to drug loading, the liposomes were prepared by hydrating a thin lipid film with a 0.3 M (NH4)2SO4 solution and reducing the size by sonication. The remote loading of Triapine, 1 and 2 was performed by mixing the drug with the liposomes for 1.5 h at 65 °C. Afterwards unloaded drug was removed by size exclusion chromatography (Sephadex G50).
The obtained liposomes were characterized by their size and morphology, PDI, EE, reproducibility as well as their size stability (Table 1). All liposomes had an average size of 50 ± 2 nm and a PDI < 0.15 indicating a narrow size distribution (Fig. 1). The zeta potential for all liposomes was measured at pH 7.4 with a charge close to neutral (ξ = 0 ± 1 mV). Furthermore, they were highly reproducible and stable in size for at least 100 days. In addition, DLS measurements revealed no changes in size after incubation of the liposomes at 37 °C for 24 h.
| Encapsulated drug | Average size (nm) of the liposome | PDI | Encapsulation efficiency (%) |
|---|---|---|---|
| Triapine | 47 ± 1 | <0.15 | 19 ± 9 |
| 1 | 55 ± 1 | <0.15 | 52 ± 5 |
| 2 | 50 ± 1 | <0.15 | 52 ± 4 |
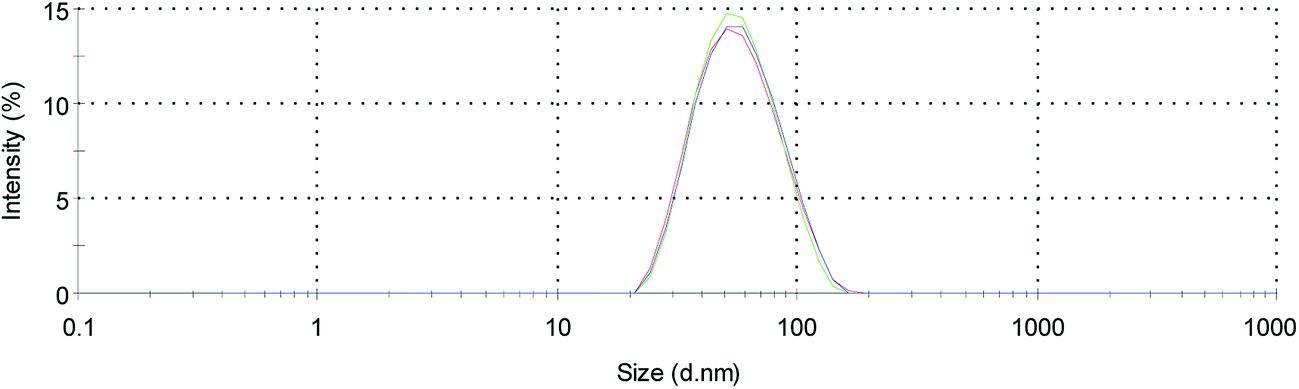 | ||
| Fig. 1 Size distribution of LP-2 measured by dynamic light scattering. | ||
Negative stain transmission electron microscopy (TEM) measurements were carried out to investigate the morphology of the liposomes. Fig. 2 shows a representative micrograph of LP-2, revealing that the liposomes have a spherical shape and confirming the size distribution measured by DLS.
 | ||
| Fig. 2 Transmission electron microscopy (TEM) image of LP-2, negatively stained with 2.5% gadolinium acetate solution. | ||
Overall, the EE of 19 ± 9% was very low for Triapine-loaded liposomes (LP-Triapine) and varied distinctly. In comparison, liposomes of 1 and 2 reached twice the drug content of LP-Triapine with ∼50% encapsulation.
Drug release from the liposomes
The drug release profile of the three thiosemicarbazone-loaded liposomes was investigated at 37 °C for 72 h (Fig. 3). For LP-Triapine, a fast burst release with more than 80% of free Triapine within the first hour was observed (Fig. 3B). For LP-1, the release was distinctly slower compared to LP-Triapine. However, still ∼40% of the free drug was released after one hour. This trend continued with ∼80% release after 48 h. In contrast, LP-2 showed controlled release behavior for more than one day without any critical burst release and less than 30% of free drug after 24 h. | ||
| Fig. 3 In vitro release profiles of LP-Triapine, LP-1 and LP-2. (A) Over 72 h with the respective best fitting release model (highest r2 value) is shown with a dashed line: first order model in case of LP-Triapine and Higuchi model in case of LP-2. (B) Zoom of the release data within the first 6 hours. | ||
To determine the in vitro release kinetics of the drugs from the liposomes, zero order, first order, and Higuchi kinetic models were applied to the obtained profiles.29 Statistical evaluation showed that LP-Triapine followed a dissolution release kinetic of the first order (with r2 values of 0.92), while LP-2 was best explained by the Higuchi model (with r2 = 0.98), which is indicative for continuous drug release by diffusion.30 None of the tested kinetic models were suitable for the description of LP-1.
Overall, this indicates that comparable to the polymeric nanoparticles, there was no stable entrapment of Triapine inside the liposomes. There are two possible explanations for this observation. On the one hand, as already mentioned above, Triapine is characterized by a rather low pyridine pK1, which might result in insufficient protonation of the drug at the pH of ∼5 inside the liposomes. Therefore, the drug might not be effectively trapped and, consequently, still able to pass the lipid membrane.26 On the other hand, our mathematical modelling indicated that LP-Triapine followed a dissolution release kinetic of first order. This suggests that Triapine is not encapsulated inside but just adsorbed to the surface of the liposomes31 due to the absence of significant drug–lipid interactions.32
In contrast to Triapine, 2 was successfully and stably encapsulated inside the liposomes. The respective drug release kinetic followed the Higuchi model (burst release followed by sustained release), which can be explained by part of the drug adsorbed to the shallow surface, with most of the drug encapsulated inside the aqueous core.33 This enhanced entrapment efficacy of 2 in comparison to Triapine is very likely linked to the introduction of an additional protonatable moiety into the thiosemicarbazone molecule, which results in better stabilization of the drug inside the liposome. In general, nanoformulations following such a diffusion-controlled release kinetic can be considered of sufficient stability for subsequent testing in vivo.31,33
Cytotoxicity and cellular drug uptake of thiosemicarbazone-loaded liposomes against human cancer cells
As a next step, the growth inhibitory effects of the novel liposomes in comparison to the free drugs were studied by MTT assays in SW480 and A2780 cells (Table 2 and Fig. 4). Noteworthy, due to their PEGylation, our liposomes can be considered as so-called “stealth liposomes”, as the PEGylated surface is known to hamper endocytic uptake via macrophages. Consequently, this kind of liposome is usually characterized by a distinctly reduced activity in cell culture compared to the free drug.34 In good agreement with our drug release data, neither LP-Triapine nor LP-1 showed any significant difference compared to the free drug. In contrast, LP-2 showed a more than 10-fold lower cytotoxicity in SW480 and 12-fold lower activity in A2780 cells indicating a stable liposomal drug formulation. Therefore, Triapine as well as 1 did not have sufficient drug loading stability for biological use, since the therapeutic benefits of liposomal formulations depend on the ability to retain the drug in the carrier and deliver it to the diseased site.19 Consequently, only LP-2 was chosen for further investigations, based on its loading capability, enhanced release characteristics as well as favorable decrease in cytotoxicity.| IC50 (μM) ± SD (μM) | ||||
|---|---|---|---|---|
| SW480 | A2780 | |||
| Triapine | 0.7 ± 0.02 | 0.7-Fold | 0.6 ± 0.01 | 0.7-Fold |
| LP-Triapine | 0.5 ± 0.03 | 0.4 ± 0.01 | ||
| 1 | 8.2 ± 0.30 | 1-Fold | 7.2 ± 0.09 | 0.9-Fold |
| LP-1 | 8.2 ± 0.41 | 6.5 ± 0.22 | ||
| 2 | 2.7 ± 0.16 | 10.2-Fold | 2.5 ± 0.16 | 12-Fold |
| LP-2 | 27.6 ± 7.75 | 29.5 ± 5.25 | ||
 | ||
| Fig. 4 Representative dose–response curves of SW480 and A2780 cells treated with 2 and LP-2. Cell viability was determined by MTT assay after 72 h treatment. Values given are means ± SD of one representative experiment performed in triplicate. | ||
To evaluate whether there is any active or passive uptake of LP-2 into cancer cells, we synthesized fluorescence-labeled liposomes loaded with 2 (NBD-LP-2) using 1% of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (NBD-DSPE). In accordance with the PEGylated nature of our liposomes, cellular uptake of NBD-LP-2 was, in general, very low. Consequently, after one hour of incubation, NBD liposomes were detectable only by the very sensitive flow cytometry (Fig. 5A) but not by fluorescence microscopy where longer incubation time (up to 18 h) was necessary to allow sufficient drug uptake for detection (Fig. 5B). However, the detected fluorescence (in flow cytometry) was significantly reduced (to ∼50%) at 4 °C (in comparison to 37 °C), indicating some active uptake of the liposomes via endocytosis. This is also in line with the vesicular localization of the fluorescence after 24 h of drug incubation (Fig. 5C).
 | ||
| Fig. 5 Cellular uptake of fluorescence (NBD)-labeled LP-2. (A) SW480 and A2780 cells were treated with NBD-LP-2 (8 μM referred to NBD) for 1 h at 37 °C or 4 °C, washed once with PBS and analyzed by flow cytometry for fluorescence intensity. *** p < 0.001 one-way ANOVA with Bonferroni posttest. (B) Fluorescence microscopy of time-dependent liposome uptake in SW480 cells treated with NBD-LP-2 (33 μM referred to NBD) for 1, 6 and 18 h at 37 °C. (C) Fluorescence microscopy of long-term liposomal uptake in SW480 cells after treatment with NBD-LP-2 (33 μM) for 24 h at 37 °C. After fixation with 4% paraformaldehyde, nuclei were stained with DAPI. Micrographs were taken with a fluorescence microscope in differential interference contrast (DIC), green (liposomes) and UV (nuclei) channels. Size bar = 20 μm. | ||
The reason for a delayed uptake of NBD-LP-2 might be the PEGylation of the liposomal surface. PEGylation is used to increase the stability of the liposomes and prolong the circulation in the bloodstream but also minimize the opsonization and thus, decrease the immediate uptake by macrophages.35 Nevertheless, partial uptake of PEGylated liposomes by cancer cells was described as well.36
Anticancer activity of LP-2
As a next step, tolerability and anticancer activity of LP-2 was evaluated in comparison to unbound 2 in CT-26-bearing Balb/c mice. As shown in Fig. 6A, all treatment schemes were well tolerated with no significant change in body weight (tested by 2-way ANOVA). With regard to the anticancer activity, 2 was significantly but only transiently reducing the tumor growth, when calculated until the last day of treatment (day 14; p < 0.001 by 2-way ANOVA; Fig. 6B). Consequently, treatment with 2 had no impact on overall survival of the treated mice (Fig. 6C). Interestingly, LP-2 had less impact than 2 on tumor growth during the two weeks of therapy. However, half of the animals reacted with tumor stabilization or shrinkage after some lag time (Fig. 6B indicated by small black arrows). This indicates that the nanoformulation LP-2 either needs some time for accumulation of sufficient intratumoral drug levels or for sufficient drug release from the liposomes. Noteworthy, it is well known that Triapine is characterized by a very low plasma-half life (less than one hour12,37,38), which together with the rather transient nature of its mode of action (stop of DNA synthesis), probably explains its lack of activity in clinical trials against solid cancer types. We have recently shown that Triapine has also strong anticancer activity against CT-26 isografts.12 However, already a therapy break of one day was sufficient to allow significant regrowth of the tumors. Due to the lack of impact of unbound 2 on the overall survival of CT-26-bearing mice, it can be expected that also this thiosemicarbazone has only a transient activity. Consequently, our experiments indicate that liposomal encapsulation indeed is a promising strategy to overcome the limitations observed in therapy with thiosemicarbazones. | ||
| Fig. 6 Tolerability and anticancer activity of LP-2vs.2in vivo. Murine CT-26 cells (5 × 105) were injected subcutaneously into the right flank of female Balb/c mice (n = 4 per group). Animals were treated with the drug intravenously (4 mg kg−1 in PBS) on day 3–7 and 10–14. (A) Body weight during therapy; (B) tumor size of the individual mice was assessed regularly by caliper measurement. Tumor volumes were calculated by using the formula: length × width2/2. Days of tumor regression are indicated by black arrows. (C) Plot of the overall survival. | ||
Methemoglobin formation
A common side effect in clinical phase I and II trials with Triapine is the oxidation of oxyhemoglobin (HbO2) to methemoglobin (metHb), the so-called methemoglobinemia.39 The occurrence of this oxidation reaction is explained by the formation of FeIII complexes of Triapine in the blood stream and subsequent oxidation of hemoglobin.40 This prompted us to study, on the one hand, if also 2 promotes formation of metHb in the presence of FeIII and, on the other hand, whether liposomal formulations of 2 are able to prevent this reaction. Hence, 2 and LP-2 were incubated with Fe(NO3)3 (2:1 ratio) for 20 min and subsequently the reaction of HbO2 was monitored by UV-Vis spectroscopy (Fig. 7). In case of the free compound 2, a distinct decrease in the absorption of the typical HbO2 bands at 577 and 539 nm accompanied with an increase at 404 and 630 nm and a hypsochromic shift of the band at 404 nm were observed after incubation with FeIII (Fig. 7A and C), indicating the fast formation of metHb.41,42 Quantitative evaluation revealed that 60 ± 3% of the total Hb are oxidized to metHb. Noteworthy, after incubation with FeIII, LP-2 shows only very small changes in the intensity of the HbO2 bands indicating negligible formation of metHb (5 ± 1% of total Hb see Fig. 7B and C). This suggests that the liposomal formulation either inhibits the formation of the redox-active FeIII complex or sterically prevents interaction of 2 with HbO2 due to the lipid barrier. 2 without Fe(NO3)3 had a much smaller effect on the HbO2 spectra (Fig. S5†).
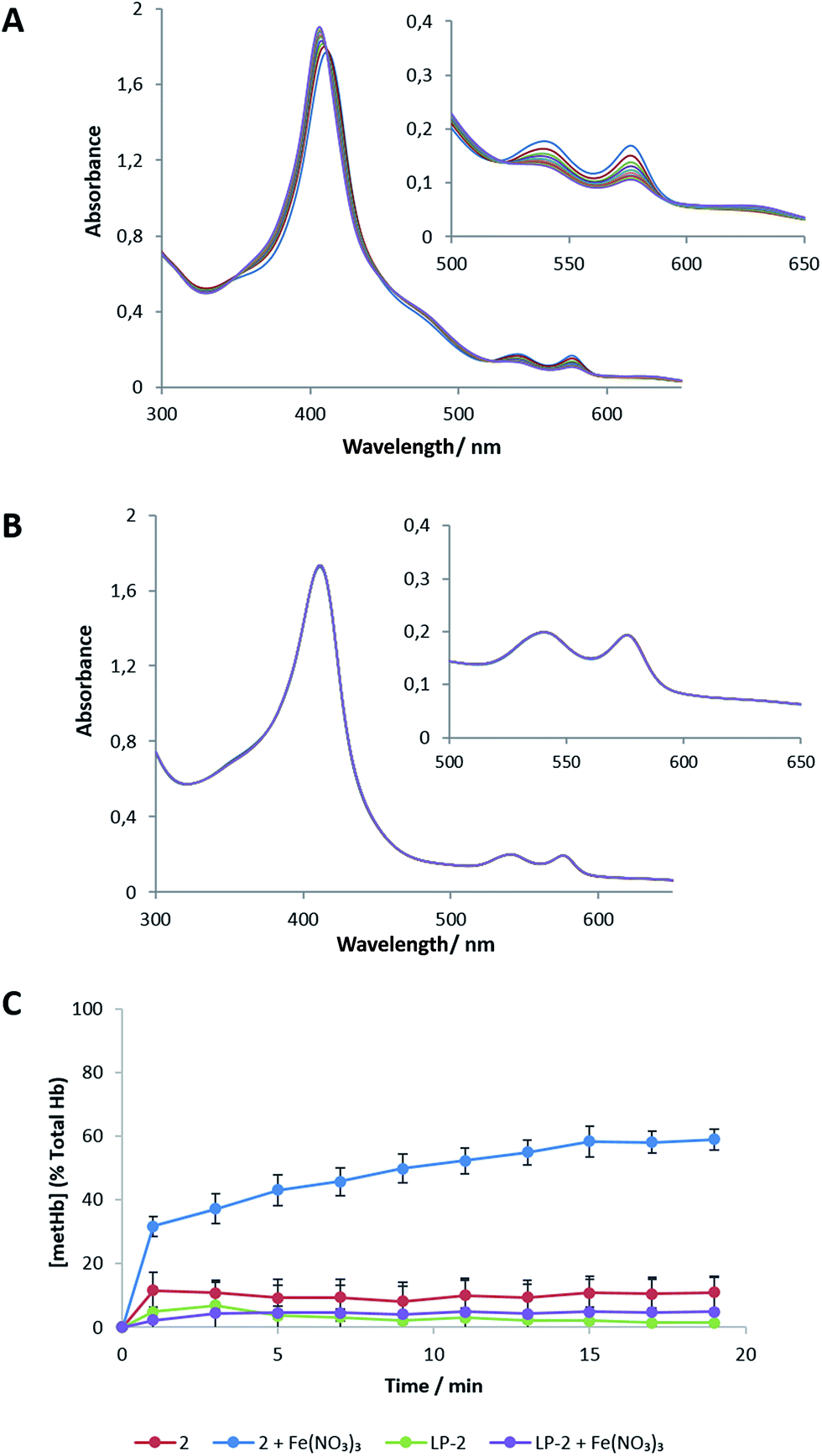 | ||
| Fig. 7 Time resolved UV-Vis spectral changes of the reaction between human HbO2 and (A) 2 in the presence of FeIII as well as (B) LP-2 in the presence of FeIII and (C) quantitative illustration of the metHb formation in the presence and absence of FeIII (results are mean ± SD (three experiments)). | ||
To assess the metHb generation of 2 and LP-2in vivo, mice were treated with a single intravenous injection of 2 or LP-2 (compared to an untreated control) and blood was collected after 60 min. The results showed the same tendency as observed in the in vitro assay (Fig. S6†), however the differences were not significant, most likely due to the limited drug concentration of the liposomal formulation, which did not allow further increase of the applied dose of both 2 and LP-2.
Conclusion
The missing activity of Triapine against solid cancer types is believed to rely on a short plasma half-time, a rapid elimination and resulting insufficient drug levels in the solid tumor tissue. Accordingly, drug delivery systems based on nanoparticles offer the opportunity to overcome these limitations by enhancement of plasma half-time and increased drug accumulation in the malignant tissue due to the so called EPR-effect. Overall, for a successful clinical development of nanocarriers, several parameters have to be considered. Besides the size, EE and reproducibility, a balanced drug release profile has a high impact on the therapeutic effectiveness, as the drug is, on the one hand, inactive while encapsulated and, on the other hand, rapidly excreted when released too fast. In view of that, this study aimed to develop a new nanoparticulate formulation of Triapine with improved delivery properties for in vivo applications.Therefore, the first Triapine-loaded polymeric nanoparticles as well as remotely loaded liposomes were successfully prepared. However, in both cases, a fast burst release was observed, which also resulted in similar cytotoxic activity compared to the free drug, which might explain, why there are no literature reports on Triapine-loaded nanoparticles so far. Consequently, two novel Triapine derivatives were synthesized and, for compound 2, a stable liposomal formulation with desired continuous drug release was achieved. By this approach, not only a significant increase in survival, but also complete prevention of methemoglobin formation, a frequently observed side effect of thiosemicarbazone treatment in clinical trials, was accomplished. In conclusion, our study shows that nanoformulations of thiosemicarbazones are a promising strategy to improve the performance of thiosemicarbazones in clinical evaluations.
Experimental section
Chemicals
PLA (MW 75000–120000), PLGA RG 503H (MW 24000–38000; Sigma Aldrich), Tween 80 (MW 1310; Fisher Scientific, Austria) and methanol (HPLC grade, Fisher Scientific, Austria) were used as supplied. 1,2-Distearoyl-sn-glycero-3-phosphocholine (DSPC), cholesterol, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt) (DSPE-mPEG(2000)) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt) (NBD-DSPE) were purchased from Avanti Polar Lipids Inc. (Alabaster, AL). Triapine was synthesized according to literature procedures.43 Human hemoglobin was purchased from Sigma Aldrich. MilliQ water was obtained from a Millipore Advantage A10 185 UV Ultrapure Water System (18.2 MΩ; Molsheim, France). All other chemicals were of analytical grade and used without further purification.
Preparation of drug-loaded nanoparticles
For preparation of the nanoparticles the nanoprecipitation method was used.27 Therefore, 40 mg of PLA (∼0.44 μmol) or PLGA (∼1.3 μmol) and 4 mg Triapine were dissolved in 3.9 mL acetone and 0.1 mL dichloromethane. This solution was poured under stirring into 10 mL of a 0.25% (w/v) aqueous phase of Tween 80 and stirred for 30 min at rt. The organic solvents were evaporated under reduced pressure at 35 °C. Free Triapine was removed by dialysis against water (MWCO: 3.5 kDa) for 24 h and the volume was adjusted under reduced pressure to 1 mL. The nanoparticles were stored at 4 °C. Drug-free nanoparticles were prepared by the same procedure.Synthesis of compound 1 and 2
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 2.87 (q, 2H, NHCH2(CH2)2), 2.49 (2H, CH2NH2, partially below the DMSO signal), 1.37–1.27 (m, 13H, C(CH3)3 and CH2(CH2)2CH2) ppm.
O), 2.87 (q, 2H, NHCH2(CH2)2), 2.49 (2H, CH2NH2, partially below the DMSO signal), 1.37–1.27 (m, 13H, C(CH3)3 and CH2(CH2)2CH2) ppm.
General procedure for the synthesis of 1b and 2b
The amine and 1 eq. of Et3N were dissolved in EtOH and 1 eq. of CS2 in EtOH was added drop-wise. The reaction mixture was stirred at rt for 1.5 h, treated with 1 eq. of CH3I and further stirred for 1.5 h. The solvent was removed under reduced pressure. EtOAc was added and the solution was extracted with 1 M HCl, saturated NaHCO3 and water. The organic layer was dried over Na2SO4, filtered and evaporated under reduced pressure and dried in vacuo.
1H NMR (DMSO-d6, 500.10 MHz): δ = 9.80 (bs, 1H, NHCS), 3.69 (t, 3J = 7 Hz, 2H, NHCH2), 2.52 (2H, CH2N(CH3)2, 3H, CH3S, partially below the DMSO signal), 2.20 (s, 6H, N(CH3)2), ppm.
S), 6.82–6.73 (m, 1H, NHCO), 3.60–3.52 and 2.95–2.88 (m, 4H, CH2(CH2)2CH2), 2.52 (3H, CH3S, partially below the DMSO signal), 1.58–1.50 (m, 2H, CH2), 1.42–1.32 (m, 11H, C(CH3)3 and CH2) ppm.
O), 4.43 (s, 2H, NH2), 3.46–3.39 and 2.95–2.89 (m, 4H, CH2(CH2)2CH2), 1.52–1.43 (m, 2H, CH2), 1.43–1.32 (m, 11H, C(CH3)3 and CH2) ppm.
N), 8.05 (d, 3J = 5.2 Hz, 1H, Hpy), 7.84 (d, 3J = 9 Hz, 1H, Hpy), 7.67 (dd, 3J = 9 Hz, 4J = 5 Hz, 1H, Hpy), 4.13 (t, 3J = 6 Hz, 2H, NHCH2), 3.52 (t, 3J = 6 Hz, 2H, CH2N(CH3)2), 3.00 (s, 6H, N(CH3)2) ppm. 13C NMR (D2O, 125.75 MHz): δ 180.0 (CS), 145.9 (Cq,py), 135.4 (NCH), 132.1 (Cpy), 131.1 (Cpy), 126.7 (Cpy), 126.3 (Cq,py), 56.4 (CH2–NH), 43.3 (N(CH3)2), 39.2 (NH–CH2–) ppm.
N), 7.99 (dd, 3J = 4 Hz, 4J = 1 Hz, 1H, Hpy), 7.78 (d, 3J = 9 Hz, 1H, Hpy), 7.60 (dd, 3J = 9 Hz, 4J = 5 Hz, 1H, Hpy), 3.67 (t, 3J = 6 Hz, 2H, NHCH2), 3.01 (t, 3J = 7 Hz, 2H, CH2NH), 1.76–1.66 (m, 4H, CH2(CH2)2CH2) ppm. 13C NMR (D2O, 125.75 MHz): δ 176.5 (CS), 145.7 (Cq,py), 134.5 (CHN), 132.1 (Cpy), 131.1 (Cpy), 126.6 (Cpy), 126.5 (Cq,py), 43.6 (NHCH2), 39.2 (CH2NH2), 25.4 ((–CH2–)), 24.0 ((–CH2–)) ppm.
Preparation of drug-loaded liposomes
Liposomes were prepared using the remote loading approach.26 Briefly, 20 mg DSPC, 6.5 mg PEG-2000PE and 7.2 mg cholesterol were dissolved in 5 mL CHCl3/MeOH (4:1, v/v) and stirred for 1.5 h at 65 °C. The solvent was removed under reduced pressure to get a thin film which was dried in vacuo overnight. The thin film was hydrated in 2 mL of a 0.3 M (NH4)2SO4 solution and 15 glass beads were added. The mixture was rotated on a rotary evaporator for 1.5 h at 65 °C. The suspension was decanted and refilled to 2 mL by 0.3 M (NH4)2SO4 solution. The suspension was homogenized by sonication with a Bandelin ultrasonic homogenizer HD 3100 with maximum amplitude of 20% for 3 min, 5 min, 7 min, 5 min and 12 min (each with few seconds break). Remaining ammonium sulfate was removed by size exclusion chromatography (Sephadex G50) using 0.1 M PBS-solution as eluent. The solid drug was doused with the purified liposomes and stirred for 1.5 h at 65 °C. Non-encapsulated drug was removed by another size exclusion chromatography (Sephadex G50; eluent: PBS). The liposomes were stored at 4 °C until further usage. Under these storage conditions, the liposomes were stable for more than 3 months, without any change in activity (data not shown).
For cell uptake studies, fluorescent-labeled liposomes (loaded with 2) were prepared as described above and 0.43 mg NBD-DSPE was added before the preparation of the thin film. The concentration used of these liposomes is referred to the attached fluorophore (0.12 mM).
Particle size and surface charge (zeta-potential)
The particle sizes and the polydispersity indices (PDI) were determined by dynamic light scattering (DLS) using a Malvern ZetaSizer Nano ZS (Malvern Instruments Ltd., Malvern, UK) equipped with a 4 mW He–Ne, 632.8 nm laser beam at 25 °C and at a scattering angle of 173°. Prior to the particle size measurement, the nanoparticles were diluted with (1:9 v/v) Millipore water, liposomes were diluted (1:9 v/v) with PBS and both measured in disposable cuvettes (UV-cuvette micro, Brand GmbH + Co KG, Germany). The zeta-potential was determined using the same instrument in disposable capillary cells.
TEM measurements
Liposomes were analyzed with negative stain electron microscopy using a Zeiss TEM 902 electron microscope. One drop (approximately 10 μL) of the diluted liposome suspension (1:9 v/v, with Millipore water) was placed on a carbon-coated 100-mesh copper grid. Any excess fluid was removed with a filter paper. The grid was allowed to dry overnight. A 2.5% gadolinium acetate solution was used as staining agent and, after 1 min, the excess was drawn off with a filter paper and the sample analyzed under the electron microscope.
Determination of the encapsulated drug amount
Aliquots of 50 μL of the nanoparticle/liposome containing solutions were dried under reduced pressure by a rotary evaporator. In case of nanoparticles the thin film was dissolved in acetone:dichloromethane (1:1, v/v) and in case of liposomes in PBS:MeOH (1:1, v/v) and sonicated for 5 min. The amount of encapsulated drug was determined by UV-Vis spectroscopy on an Agilent 8453 UV-Visible system (Agilent Technologies, Germany) using a calibration curve of the drug. The samples were measured in 10 mm path length quartz cuvettes.
Stability and reproducibility of particle size
Three batches of freshly prepared nanoparticles and liposomes were investigated to determine the reproducibility in size and drug loading. Therefore, the mean diameter and PDI of three independent batches was measured by DLS and the drug content was analyzed by UV-Vis spectroscopy as described above. Furthermore, long-term stability studies over a 100 days and heat stability at 37 °C for 24 h were carried out by determination of the particle sizes with DLS as described above.Drug release studies
The dialysis bag diffusion technique was used to study the drug release. Drug-loaded particles were freshly prepared and added into a dialysis membrane (average flat width of 10 mm and molecular weight cut off of 12.5 kDa (Sigma Aldrich, Austria)). The membrane was closed and immersed into a 0.1 M phosphate-buffered saline (PBS) solution pH = 7.4 at 37 ± 1 °C with continuous stirring at 200 rpm. To determine the amount of diffused drug through the dialysis membrane, 1 mL samples were withdrawn from the solution at fixed time intervals (0 min, 1 h, 3 h, 6 h, 24 h, 48 h and 72 h) and replaced by fresh PBS solution. The drug concentration was measured by fluorescence spectrophotometry (Horiba Jobin Yvon, FluoroMax 4) at rt (excitation wavelength 368 nm/emission wavelength 455 nm). As positive control stock solutions of the free drug in DMSO were diluted to the concentration of the drug in the nanoformulations by 0.1 M PBS (final DMSO concentration ≤ 1%). The solution was placed into the dialysis bag and the drug content in the outer compartment was determined as described for the nanoformulations. The maximum value of released free drug was set to 100%.Subsequently, release profiles of liposomal formulations were tested for their suitability to zero order, first order and Higuchi models29 by the determination of R2 value using GraphPad Prism software.
Methemoglobin formation
Prior to kinetic measurements, fresh solutions of HbO2 were prepared by dissolving human hemoglobin (ca. 40 mg) in 2 mL of 0.1 M PBS and reducing the metalloprotein by incubation with an excess of sodium dithionite (ca. 5 mg) for 1.5 h at rt. The resulting solution was purified by gel filtration (Sephadex G-25) and PBS as eluent. The concentration of HbO2 was determined from the extinction coefficient of the protein at 542 nm.41 Stock solutions of 2 (5 mM) and Fe(NO3)3 (50 mM) were prepared in MeOH. LP-2 and 2 were incubated with Fe(NO3)3 in a 2:1 molar ratio for 10 min at rt and diluted with 0.1 M PBS (maximum MeOH content did not exceed 1%). For kinetic studies, the incubated solutions (final concentration referred to 2: 50 μM) and HbO2 (final concentration: 5 μM total heme) in PBS were measured over 20 minutes using a Perkin Elmer UV-Vis spectrometer Lambda 35 over 10 cycles (300–650 nm; scan-speed 480 nm min−1). 2 and LP-2 (as negative control) were measured without the prior incubation with Fe(NO3)3 (final concentration HbO2 5 μM total heme and drug 50 μM). For quantification the concentrations of HbO2 and metHb were determined at 577 and 630 nm after subtraction of the controls (HbO2 or the compound alone at 630 nm) from the extinction coefficient.41
To assess the metHb formation in vivo mice were treated intravenously with 2 or LP-2 (drug concentration each 3 mg kg−1). After 60 min, mice were anesthetized and blood collected by heart puncture in a heparine tube. Lysis of red blood cell compartment was performed as previously published.39 Quantification of HbO2 and metHb levels was performed as before.
Cell culture
The human colon cancer cell line SW480 was from ATCC (VA, USA) and grown in MEM growth media. The human ovarian cancer cell line A2780 was purchased from Sigma-Aldrich (MO, USA) and grown in RPMI-1640 growth media. The murine colon cancer cell line CT-26 (from ATCC) was grown in DMEM/F12 medium (Sigma). Culture growth media were supplemented with 10% fetal calf serum (FCS) (PAA, Pasching, Austria). Cell cultures were grown and maintained in a cell incubator (37 °C, 5% CO2, and humidified atmosphere) and periodically checked for mycoplasma contamination. Authentication of cell models was performed by short tandem repeat (STR) fingerprint.Cell viability assay
Activity of the investigated compounds was tested in 2 to 4 × 103 cells per well seeded in 96-well plates. After 24 h recovery, cells were treated with the investigated compounds diluted in respective culture media at indicated concentrations and incubated for 72 h. Cell viability was measured with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)-based vitality assay (EZ4U; Biomedica, Vienna, Austria) following the manufacturer's recommendations. IC50 values were calculated from dose response curves using GraphPad Prism software (La Jolla, USA).Uptake of liposomal particles
The uptake of NBD-labeled liposomes (NBD-LP-2) loaded with 2 was measured by flow cytometry and fluorescence microscopy. For flow cytometry, 2–4 × 105 cells per well were seeded in 6-well plates 24 h prior to treatment. Cells were incubated with NBD-LP-2 (end concentration 7 μM NBD) at 37 °C and 4 °C for 1 h. After incubation, cells were washed twice with ice-cold PBS and detached with trypsin. Cells were washed twice with ice-cold PBS and immediately measured using a BD LSRFortessa™ Flow cytometer (BD Biosciences, NJ, USA). Total fluorescence was further analyzed using Flowing Software (University of Turku, Finland).For microscopy experiments, 3 × 105 cells were seeded in ibidi 15 μ slides (8-well) with 180 μm polymer coverslips (Martinsried, Germany). After incubation with NBD-LP-2 (end concentration 33 μM NBD) for 1, 6, 18 and 24 h, cells were washed with ice-cold PBS, fixed with 4% paraformaldehyde (PFA) for 20 min, washed with ice-cold PBS and finally stained with 0.5 μg mL−1 DAPI for 5 min. After washing with distilled/deionized H2O, cells were acquired in differential interference contrast (DIC), DAPI (blue) and NBD (green) channels using an inverted microscope (Nikon Eclipse Ti, Life-Cell Imaging from Visitron Systems, Germany).
Animal experiments
All animal experiments were approved by the local ethics commission and carried out according to the Austrian and FELASA guidelines for animal care and protection. Six- to eight-week-old female Balb/c mice (weighing ∼ 20 g) were purchased from Harlan Laboratories, San Pietro al Natisone, Italy. The animals were kept in a pathogen-free environment, and every procedure was done in a laminar airflow cabinet.Murine CT-26 cells (5 × 105) were injected subcutaneously into the right flank of female Balb/c mice. Animals were intravenously treated with the drugs (4 mg kg−1 dissolved in PBS) on days 3–7 and 10–14. Animals were controlled for distress development every day, and tumor size was assessed regularly by caliper measurement. Tumor volume was calculated using the formula: length × width2/2.
Acknowledgements
We thank Mag. Gerhard Zeitler for devoted animal care and Thomas Mohr for help with the release kinetic models. Daniela Gruber of the Core Facility Cell Imaging and Ultrastructure Research of the University of Vienna for introduction and help concerning the TEM measurements. This work was supported by the exploratory focus “Functionalized Materials and Nanostructures” of the University of Vienna, the Austrian Science Fond (FWF) grant P22072 (to W. Berger), the Mahlke geb. Obermann-Stiftung of the University of Vienna FA526009 (to B. Keppler) and was performed with the help of the COST action CM1105.References
- M. Kolberg, K. R. Strand, P. Graff and K. K. Andersson, Biochim. Biophys. Acta, Proteins Proteomics, 2004, 1699, 1–34 CrossRef CAS PubMed.
- D. J. R. Lane, T. M. Mills, N. H. Shafie, A. M. Merlot, R. Saleh Moussa, D. S. Kalinowski, Z. Kovacevic and D. R. Richardson, Biochim. Biophys. Acta, Rev. Cancer, 2014, 1845, 166–181 CrossRef CAS PubMed.
- Y. Aye, M. J. C. Long and J. Stubbe, J. Biol. Chem., 2012, 287, 35768–35778 CrossRef CAS PubMed.
- G. Y. Lui, Z. Kovacevic, V. Richardson, A. M. Merlot, D. S. Kalinowski and D. R. Richardson, Oncotarget, 2015, 6, 18748 CrossRef PubMed.
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2013, 114, 815–862 CrossRef PubMed.
- P. J. Jansson, D. S. Kalinowski, D. J. Lane, Z. Kovacevic, N. A. Seebacher, L. Fouani, S. Sahni, A. M. Merlot and D. R. Richardson, Pharmacol. Res., 2015, 100, 255–260 CrossRef CAS PubMed.
- http://www.clinicaltrials.gov .
- F. J. Giles, P. M. Fracasso, H. M. Kantarjian, J. E. Cortes, R. A. Brown, S. Verstovsek, Y. Alvarado, D. A. Thomas, S. Faderl, G. Garcia-Manero, L. P. Wright, T. Samson, A. Cahill, P. Lambert, W. Plunkett, M. Sznol, J. F. DiPersio and V. Gandhi, Leuk. Res., 2003, 27, 1077–1083 CrossRef CAS PubMed.
- J. E. Karp, F. J. Giles, I. Gojo, L. Morris, J. Greer, B. Johnson, M. Thein, M. Sznol and J. Low, Leuk. Res., 2008, 32, 71–77 CrossRef CAS PubMed.
- B. Ma, B. C. Goh, E. H. Tan, K. C. Lam, R. Soo, S. S. Leong, L. Z. Wang, F. Mo, A. T. C. Chan, B. Zee and T. Mok, Invest. New Drugs, 2007, 26, 169–173 CrossRef PubMed.
- J. J. Knox, S. J. Hotte, C. Kollmannsberger, E. Winquist, B. Fisher and E. A. Eisenhauer, Invest. New Drugs, 2007, 25, 471–477 CrossRef CAS PubMed.
- K. Pelivan, W. Miklos, S. van Schoonhoven, G. Koellensperger, L. Gille, W. Berger, P. Heffeter, C. R. Kowol and B. K. Keppler, J. Inorg. Biochem., 2015 DOI:10.1016/j.jinorgbio.2015.10.006.
- C. A. Kunos, T. Radivoyevitch, S. T. Ingalls and C. L. Hoppel, Future Oncol., 2012, 8, 145–150 CrossRef CAS PubMed.
- N. Portney and M. Ozkan, Anal. Bioanal. Chem., 2006, 384, 620–630 CrossRef CAS PubMed.
- H. Maeda, H. Nakamura and J. Fang, Adv. Drug Delivery Rev., 2013, 65, 71–79 CrossRef CAS PubMed.
- B. Mishra, B. B. Patel and S. Tiwari, Nanomedicine: Nanotechnology, Biology and Medicine, 2010, 6, 9–24 CrossRef CAS PubMed.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- J. P. May, E. Undzys, A. Roy and S.-D. Li, Bioconjugate Chem., 2016, 27, 226–237 CrossRef CAS PubMed.
- I. V. Zhigaltsev, G. Winters, M. Srinivasulu, J. Crawford, M. Wong, L. Amankwa, D. Waterhouse, D. Masin, M. Webb, N. Harasym, L. Heller, M. B. Bally, M. A. Ciufolini, P. R. Cullis and N. Maurer, J. Controlled Release, 2010, 144, 332–340 CrossRef CAS PubMed.
- T. Musumeci, C. A. Ventura, I. Giannone, B. Ruozi, L. Montenegro, R. Pignatello and G. Puglisi, Int. J. Pharm., 2006, 325, 172–179 CrossRef CAS PubMed.
- K. S. Soppimath, T. M. Aminabhavi, A. R. Kulkarni and W. E. Rudzinski, J. Controlled Release, 2001, 70, 1–20 CrossRef CAS PubMed.
- E. Leo, B. Brina, F. Forni and M. A. Vandelli, Int. J. Pharm., 2004, 278, 133–141 CrossRef CAS PubMed.
- R. Mackenzie, J. Booth, C. Alexander, M. C. Garnett and C. A. Laughton, J. Chem. Theory Comput., 2015, 11, 2705–2713 CrossRef CAS PubMed.
- S. Puri, P. Kallinteri, S. Higgins, G. A. Hutcheon and M. C. Garnett, J. Controlled Release, 2008, 125, 59–67 CrossRef CAS PubMed.
- Q. Miao, D. Xu, Z. Wang, L. Xu, T. Wang, Y. Wu, D. B. Lovejoy, D. S. Kalinowski, D. R. Richardson and G. Nie, Biomaterials, 2010, 31, 7364–7375 CrossRef CAS PubMed.
- D. Zucker, D. Marcus, Y. Barenholz and A. Goldblum, J. Controlled Release, 2009, 139, 73–80 CrossRef CAS PubMed.
- É. A. Enyedy, É. Zsigó, N. V. Nagy, C. R. Kowol, A. Roller, B. K. Keppler and T. Kiss, Eur. J. Inorg. Chem., 2012, 2012, 4036–4047 CrossRef.
- É. A. Enyedy, N. V. Nagy, É. Zsigó, C. R. Kowol, V. B. Arion, B. K. Keppler and T. Kiss, Eur. J. Inorg. Chem., 2010, 2010, 1717–1728 CrossRef.
- V. Raju and K. V. Murthy, Indian J. Pharm. Sci., 2011, 73, 527–536 CrossRef CAS PubMed.
- H. Ocal, B. Arica-Yegin, I. Vural, K. Goracinova and S. Calis, Drug Dev. Ind. Pharm., 2014, 40, 560–567 CrossRef PubMed.
- S. K. Adesina, A. Holly, G. Kramer-Marek, J. Capala and E. O. Akala, J. Pharm. Sci., 2014, 103, 2546–2555 CrossRef CAS PubMed.
- H. Grohganz, I. Tho and M. Brandl, Eur. J. Pharm. Biopharm., 2005, 59, 439–448 CrossRef CAS PubMed.
- H. Zhang, F. M. Zhang and S. J. Yan, Int. J. Nanomed., 2012, 7, 2901–2910 CrossRef CAS PubMed.
- A. Hioki, A. Wakasugi, K. Kawano, Y. Hattori and Y. Maitani, Biol. Pharm. Bull., 2010, 33, 1466–1470 CAS.
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 CAS.
- Y. Sadzuka, K. Kishi, S. Hirota and T. Sonobe, J. Liposome Res., 2003, 13, 157–172 CrossRef CAS PubMed.
- L. Feun, M. Modiano, K. Lee, J. Mao, A. Marini, N. Savaraj, P. Plezia, B. Almassian, E. Colacino, J. Fischer and S. MacDonald, Cancer Chemother. Pharmacol., 2002, 50, 223–229 CrossRef CAS PubMed.
- J. Murren, M. Modiano, C. Clairmont, P. Lambert, N. Savaraj, T. Doyle and M. Sznol, Clin. Cancer Res., 2003, 9, 4092–4100 CAS.
- P. Quach, E. Gutierrez, M. T. Basha, D. S. Kalinowski, P. C. Sharpe, D. B. Lovejoy, P. V. Bernhardt, P. J. Jansson and D. R. Richardson, Mol. Pharmacol., 2012, 82, 105–114 CrossRef CAS PubMed.
- M. T. Basha, J. Bordini, D. R. Richardson, M. Martinez and P. V. Bernhardt, J. Inorg. Biochem., 2015 DOI:10.1016/j.jinorgbio.2015.12.004.
- W. G. Zijlstra, A. Buursma and W. P. Meeuwsen-van der Roest, Clin. Chem., 1991, 37, 1633–1638 CAS.
- M. T. Basha, C. Rodríguez, D. R. Richardson, M. Martínez and P. V. Bernhardt, J. Biol. Inorg. Chem., 2013, 19, 349–357 CrossRef PubMed.
- C. R. Kowol, R. Trondl, P. Heffeter, V. B. Arion, M. A. Jakupec, A. Roller, M. Galanski, W. Berger and B. K. Keppler, J. Med. Chem., 2009, 52, 5032–5043 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra07659a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |