
Effective cementation and removal of arsenic with copper powder in a hydrochloric acid system
Abstract
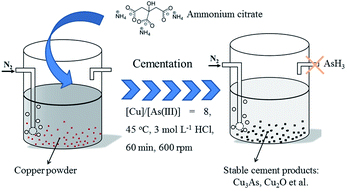
This work investigated the removal and cementation of arsenic from a hydrochloric acid system with copper powder. Thermodynamic analysis and cyclic voltammetry test were conducted to evaluate the feasibility of cementation. The effect of reaction temperature, mole ratio of Cu to As(III), stirring rate, reaction time and HCl concentration on the cementation efficiency of arsenic were investigated systematically. 94.75% of the arsenic could be removed under optimized conditions: mole ratio of Cu/As = 8 at 45 °C for 60 min. The structure and morphologies of the cement products were characterized by X-ray diffraction and scanning electron microscopy, respectively. The results show that the reaction temperature has little influence on the morphology of the cement products which consist of particles with various sizes, but has a great influence on the cementation efficiency. During the cementation process, the rate-controlling mechanisms are changed at different temperatures. It is diffusion controlled at the high temperature region (45–50 °C) with an activation energy of 26.7 kJ mol−1, while it is surface reaction controlled at the lower temperature region (30–45 °C) with an activation energy of 145.7 kJ mol−1. Ammonium citrate can efficiently inhibit the evolution of arsane, while has little influence on the cementation efficiency of arsenic.
 Please wait while we load your content...
Please wait while we load your content...

