
Heteroarm core cross-linked star polymers via RAFT copolymerization of styrene and bismaleimide†
Nam Young Ahn and
Myungeun Seo*
Graduate School of Nanoscience and Technology, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Korea. E-mail: seomyungeun@kaist.ac.kr
First published on 9th May 2016
Abstract
We explored reversible addition-fragmentation chain transfer (RAFT) copolymerization of 1,2-bis(maleimidoethane) (BMI) with styrene (S) in the presence of polylactide macro-chain transfer agent (PLA-CTA) as a means to synthesize heteroarm core cross-linked star (CCS) polymers consisting of PLA and PS arms (PLAnPSn). Because of the strong alternating tendency of maleimide and styrenic double bonds, copolymerization of BMI with an excess of S depleted BMI in the early stage of polymerization forming a cross-linked core. The remaining S was successively polymerized to grow PS arms from the core, completing PLAnPSn via “in–out” mechanism. Use of a stoichiometric amount of S produced PLAn, which could be used as a macro-CTA for the synthesis of more well-defined PLAnPSn. Compared with divinylbenzene, copolymerization of BMI with S was much more effective for core formation suggesting the importance of the alternating character of the copolymerization. While PLAnPSn existed as stable nanoparticles in a neutral solvent in contrast to linear PLA-b-PS, it also self-assembled to form microphase-separated structures in a selective solvent and in bulk indicating that PLA and PS arms can be intramolecularly segregated.
Introduction
Heteroarm or miktoarm star polymers consisting of two or more different polymer chains (arms) covalently connected to a core have attracted attention due to their unique properties originating from the architecture.1–4 Compared to the linear diblock analogues, different behaviors on microphase separation and micellization have been observed from heteroarm star polymers.5–10 A few examples have suggested that they can exhibit Janus nanoparticle-like characters when phase separation occurs within the heteroarm star polymer.11–16The “Arm-first” approach has been a popular synthetic route to heteroarm star polymers which involves cross-linking polymerization in the presence of end-functionalized polymers.3,17 As the approach produces star polymers possessing densely cross-linked cores, the resulting polymer are referred to as heteroarm core cross-linked star (CCS) polymers.17 While the “in–out” method produces heteroarm CCS polymers by polymerization of a multifunctional vinyl cross-linker such as divinylbenzene (DVB) in the presence of a macroinitiator (for example, polystyryl carbanion) which is followed by addition of the second monomer to grow new arms from the core,18 the “multi-macroinitiator” method ties different polymers to the core by polymerizing the cross-linker in the presence of different macroinitiators.19
Use of living or controlled polymerization reactions such as anionic polymerization is critical for both methods. CRP can be particularly useful for the synthesis of functional CCS polymers thanks to high functionality tolerance of CRP process.20 Polymerization of DVB via atom transfer radical polymerization (ATRP)21 or nitroxide-mediated polymerization (NMP)22 has been successfully used for formation of CCS polymers. However, DVB polymerization in the presence of macro-chain transfer agents via reversible addition-fragmentation chain transfer (RAFT) process produced polymers with broad dispersities.23–25 It appears that incorporation of linear polymers into the core via RAFT process is less effective compared with other CRPs due to the intrinsic reversible chain transfer mechanism.23 Conducting RAFT dispersion polymerization where the emergent core becomes insoluble has been suggested as an alternative route to produce amphiphilic heteroarm CCS polymers.26,27 We also note that a recent example has reported successful synthesis of CCS and heteroarm CCS polymers via “in–out” method utilizing visible light mediated RAFT photopolymerization.28
Here we present a facile method to produce heteroarm CCS polymers via RAFT polymerization process utilizing alternating copolymerization of styrene and maleimide. Controlled radical copolymerization of maleic anhydride (MA) with S in excess is known to undergo in an alternating fashion until the MA monomer is depleted, and then homopolymerization of the remaining S monomer proceeds to yield a P(S-alt-MA)-b-PS diblock polymer in one step.29,30 Furthermore, copolymerization of BMI with excess of S has been shown to form CCS polymers possessing a cross-linked P(S-alt-BMI) core and PS arms.31 Copolymerization of a different bismaleimide with S in the presence of PS macrointiator has been also systematically studied for synthesis of PS CCS polymers.32
Inspired by these works, we explored copolymerization of 1,2-bis(maleimidoethane) (BMI) with an excess of styrene (S) in the presence of polylactide macro-chain transfer agent (PLA-CTA) to synthesize heteroarm CCS polymers consisting of PLA and PS arms via the “in–out” method. We demonstrate that BMI is rapidly depleted in the early stage of polymerization to form CCS polymers with PLA arms, and then successive growth of PS from the core completes formation of the heteroarm CCS polymer. Compared to DVB which hardly produced star polymers under the identical condition, use of S/BMI dramatically accelerated formation of the core due to the alternating tendency between styrenic and maleimide double bonds. The heteroarm CCS polymer existed as nanoparticles in toluene (solvent good to both PLA and PS) but also self-assembled into supermicelles33 in acetonitrile (solvent selective to PLA34) suggesting Janus-like characters. Films of the heteroarm CCS polymers also exhibited microphase-separated morphologies supporting that PLA and PS arms can be intramolecularly segregated.
Results and discussion
The synthetic route to the heteroarm CCS polymer is depicted in Scheme 1. We denote the resulting polymer as PLAnPSn, where n represent the average number of the polymer chain (arm) per star polymer. We assumed the number of PLA and PS arms would be equal neglecting the small fraction of chains initiated by a radical initiator and any irreversible chain termination events. Under these conditions, all the PS chains should be initiated from the R group of the chain transfer agent, which is attached to the end of PLA chain.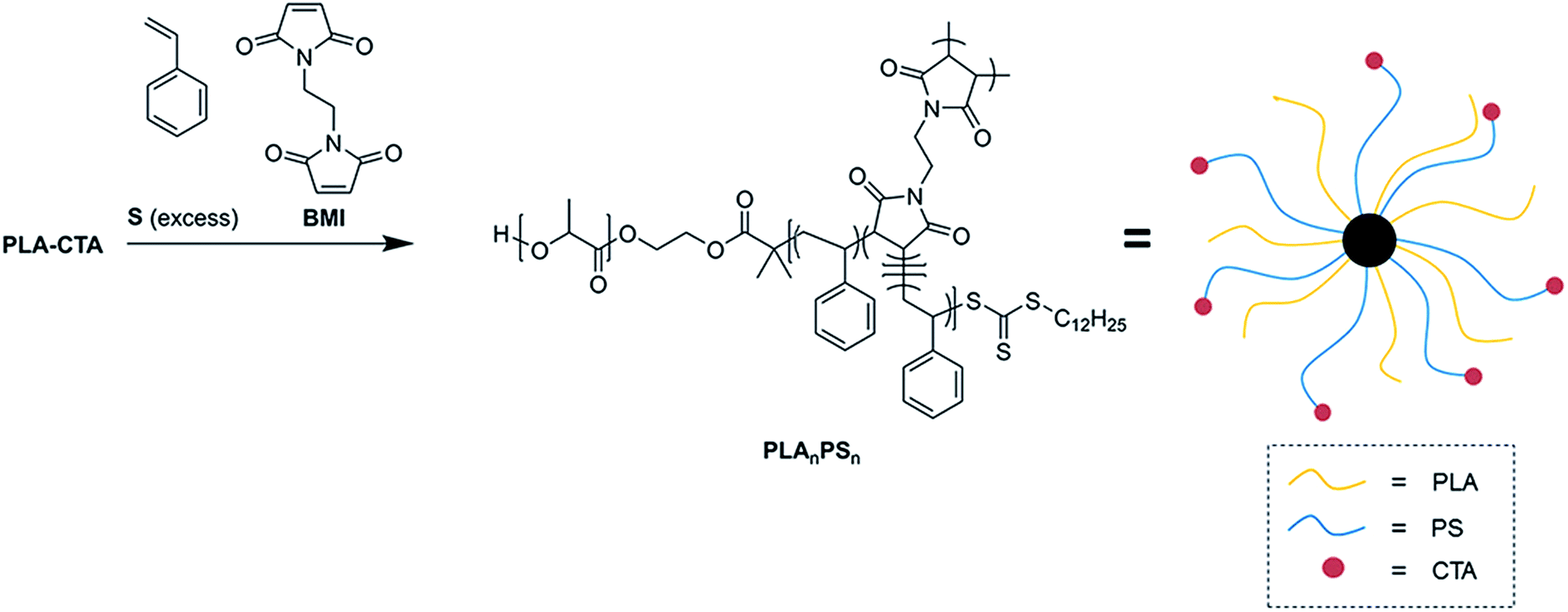 | ||
| Scheme 1 Schematic depiction of the heteroarm CCS polymer PLAnPSn. | ||
Fig. 1 and Table 1 summarize copolymerization kinetics data of BMI with excess of S obtained at 60 °C in the presence of PLA-CTA, azobisisobutyronitrile (AIBN) as a radical initiator and N,N-dimethylformamide (DMF) as a solvent. PLA-CTA with the number-average molar mass (Mn) of 30 kg mol−1 was prepared according to the literature and used (Fig. S1†).35 The molar ratio was [S]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[BMI]:[PLA-CTA]:[AIBN] = 500:8:1:0.1. 4 ampoules containing the identical polymerization mixture were individually prepared and opened after a certain time lapse of polymerization to obtain polymers by precipitating in methanol.
:[BMI]:[PLA-CTA]:[AIBN] = 500:8:1:0.1. 4 ampoules containing the identical polymerization mixture were individually prepared and opened after a certain time lapse of polymerization to obtain polymers by precipitating in methanol.
 | ||
| Fig. 1 Copolymerization of BMI with an excess of S in the presence of PLA-CTA, AIBN, and DMF at 60 °C. (a) Kinetic plot with respect to S concentration. (b) SEC traces over time. (c) Conversion vs. Mn and Đ plot. | ||
| Entry | Polymerization time (h) | Conv.a (%) | Mn,NMR,PSb (kg mol−1) | fPSc | Mn,SECd (kg mol−1) | Đd | Mw,MALLSe (kg mol−1) | Đe | nf |
|---|---|---|---|---|---|---|---|---|---|
| a Estimated by 1H NMR analysis of the aliquots.b Conversion estimated by 1H NMR analysis of the polymers based on the integration of PLA and PS protons assuming Mn,PLA = 30 kg mol−1.c Assuming the polymer is only composed of PLA and PS and their densities are 1.25 and 1.05 g mL−1, respectively.d Determined by CHCl3-SEC with a RI detector based on linear PS standards.e Determined by THF-SEC with a MALLS detector.f Estimated from Mw,MALLS. | |||||||||
| 1 | 12 | 14 | 7 | 0.22 | 342 | 1.30 | 666 | 1.8 | 11.7 |
| 2 | 24 | 22 | 11 | 0.30 | 414 | 1.32 | 1112 | 2.1 | 15.8 |
| 3 | 36 | 31 | 16 | 0.39 | 458 | 1.34 | 1580 | 2.1 | 20.5 |
| 4 | 48 | 37 | 18 | 0.42 | 485 | 1.36 | 1366 | 2.3 | 15.1 |
Consumption of S monitored by 1H nuclear magnetic resonance (NMR) spectroscopy throughout the polymerization suggested first-order kinetics consistent with the RAFT mechanism (Fig. 1a). In a separate run following the polymerization condition described above, we observed that BMI was completely consumed after 4 h of polymerization while conversion of S was 8% (Fig. S2a†). 1H NMR spectra of the produced polymers indicated that the PS mole fraction gradually increased over conversion, but the spectra were indistinguishable from linear PLA-b-PS, presumably due to the low concentration and limited mobility of BMI repeating units in the polymer (Fig. S2b and S3†). Mn (Mn,NMR,PS) and volume fraction (fPS) of PS arms were also calculated from the PS mole fraction values and listed in Table 1.
Size exclusion chromatography (SEC) traces of the polymers obtained with a refractive index (RI) detector clearly revealed that high molar mass species (>300 kg mol−1) were produced (Fig. 1b). This suggests that BMI was depleted in the early stage of the polymerization to form a cross-linked P(S-alt-BMI) core. Indeed, formation of the cross-linked core was observed even 8% conversion of S, indicating strong alternating tendency of S and BMI (Fig. S2c†). We note that the SEC traces also contained two minor peaks with smaller molar masses, presumably due to formation of linear PLA-b-PS and homoPS (Fig. S4†). An attempt to further purify PLAnPSn by reprecipitation in methanol was not successful. Molar masses of PLAnPSn calculated from the SEC traces based on linear PS standards exhibited a gradual increase as the conversion of S increased (Fig. 1c). Continued growth of PS arms after the core formation was consistent with the “in–out” mechanism. Dispersities (Đ) of PLAnPSn were maintained as ca. 1.3 throughout the polymerization, although increased slightly over conversion.
Without further purification, we estimated weight-average absolute molar mass of PLAnPSn by employing a multi-angle laser light scattering (MALLS) detector in the SEC analysis (Fig. S5†). Assuming that the number of PLA and PS arms is equal and the core is exclusively composed of P(S-alt-BMI), the average arm number n in the range of 10–20 was extracted. We posit that differences between n mainly originate from batch-to-batch variation as [BMI]:[PLA-CTA] was fixed. We also note that preliminary screening on the effect of [S]:[BMI]:[PLA-CTA] suggests the optimal molar ratio may exist for the synthesis of PLAnPSn with minimal formation of linear PLA-b-PS (Table S1 and Fig. S6†) as reported in the literature for CCS polymer synthesis by copolymerization of S and a different bismaleimide via NMP.32 Overall, all the data support formation of PLAnPSn via alternating copolymerization of BMI with S in the presence of PLA-CTA.
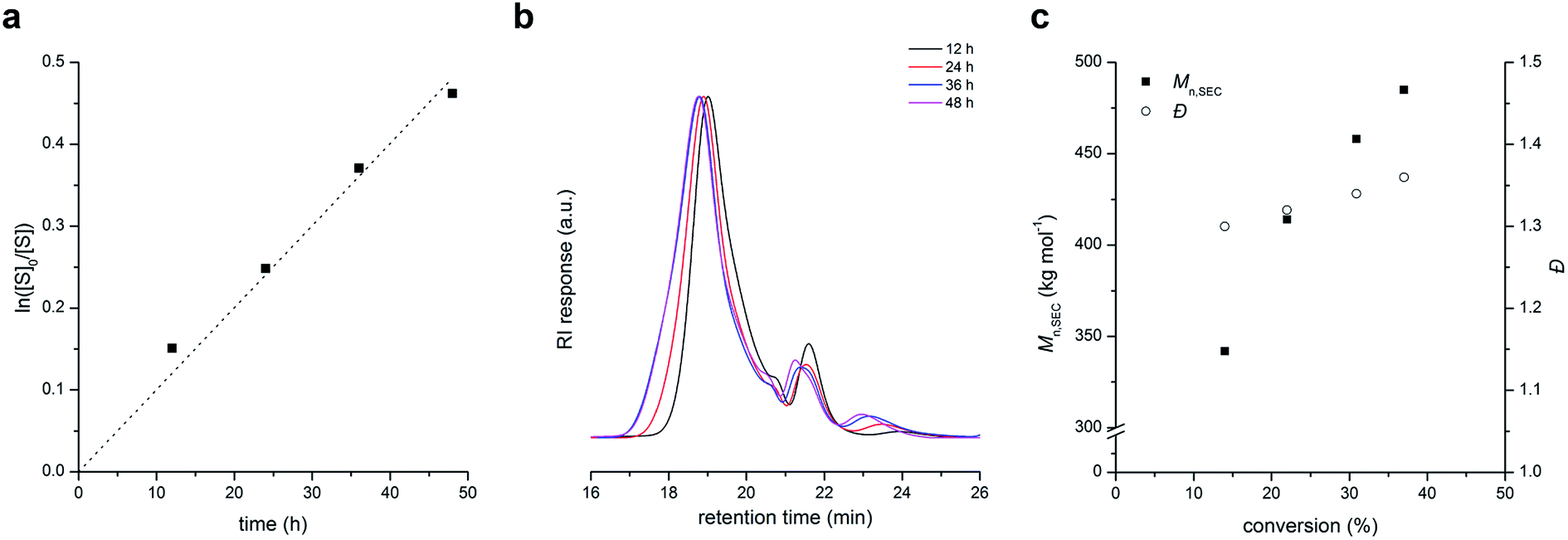
We found that the one-step alternating copolymerization of BMI with excess of S can be divided into two steps: copolymerization of stoichiometric amounts of BMI and S (i.e., 1:2 molar ratio) in the presence of PLA-CTA to produce PLAn (“in” part), and subsequent polymerization of S using the PLAn as a macro-CTA (“out” part). Fig. 2a shows an overlay of SEC traces (recorded with the RI detector) of PLAn (Mn,SEC = 401 kg mol−1 and Đ = 1.12) and PLAnPSn (Mn,SEC = 453 kg mol−1 and Đ = 1.14) obtained by sequential polymerization using PLA-CTA with Mn = 22 kg mol−1. Compared with PLAnPSn obtained by one-step alternating copolymerization, sequential polymerization appears to produce more well-defined PLAnPSn although an additional step is required. The superior control was attributed to the stronger tendency to undergo alternating copolymerization in the stage of core formation due to the equal amounts of styrenic and maleimide double bonds, which would generate PLAn minimizing incorporation of PS in the core. To achieve both high controllability and synthetic feasibility, we demonstrated one-pot process including stoichiometric copolymerization of BMI and S followed by addition of excess S into the pot after 24 h also produced well-defined PLAnPSn (Mn,SEC = 644 kg mol−1 and Đ = 1.14) (Fig. 2b).
 | ||
| Fig. 2 (a) SEC traces of PLAn (solid line) obtained by copolymerization of stoichiometric amounts of BMI and S in the presence of PLA-CTA ([S]:[BMI]:[PLA-CTA] = 16:8:1) for 24 h, and PLAnPSn (dashed line) obtained by chain extension of S using PLAn as a macro-CTA ([S]:[PLA-CTA] = 500:1). (b) SEC trace of PLAnPSn obtained by copolymerization of stoichiometric mounts of BMI and S in the presence of PLA-CTA followed by addition of S in one pot. | ||
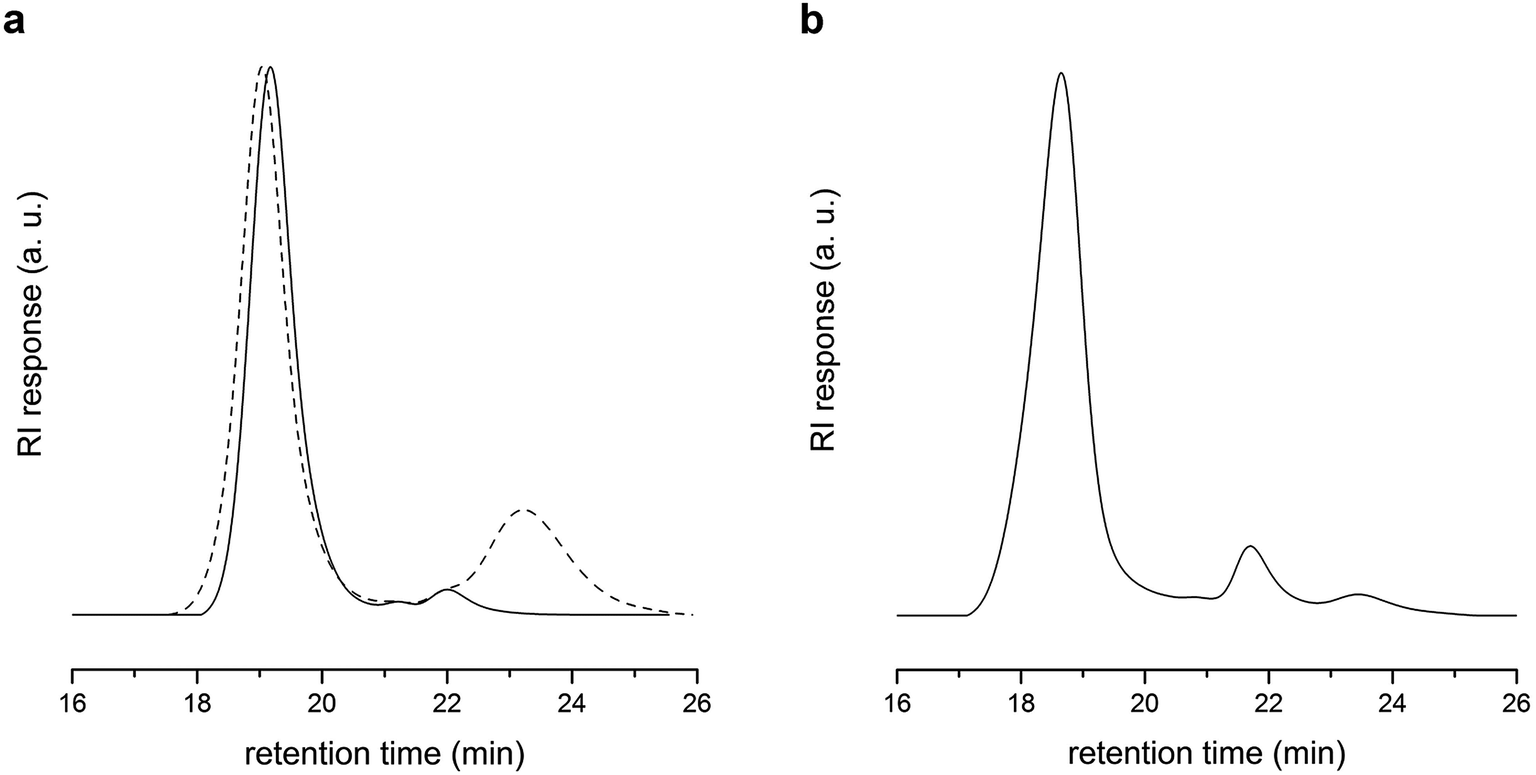
As a comparison, copolymerization of DVB with excess of S in the presence of PLA-CTA ([S]:[DVB]:[PLA-CTA] = 500:8:1) at 60 °C for 36 h resulted in insufficient coupling of PLA-CTA and mostly produced linear PLA-b-PS as evidenced by SEC analysis (Fig. 3a). Moreover, almost no polymerization proceeded when homopolymerization of DVB in the presence of PLA-CTA ([DVB]:[PLA-CTA] = 8:1) was performed for 24 h.36 This suggests that copolymerization of S and BMI via RAFT process generates a cross-linked core much more efficiently and rapidly compared with that of DVB.
 | ||
| Fig. 3 (a) SEC trace of polymer obtained by copolymerization of S and DVB in the presence of PLA-CTA ([S]:[DVB]:[PLA-CTA] = 500:8:1) (Mn,SEC = 47 kg mol−1 and Đ = 1.27). (b) SEC trace of polymer obtained by polymerization of DVB in the presence of PLA-CTA ([DVB]:[PLA-CTA] = 8:1) (Mn,SEC = 31 kg mol−1 and Đ = 1.08). | ||
Polymerization kinetics of S/BMI (stoichiometric ratio) and DVB in the presence of PLA-CTA was studied to quantitatively evaluate differences in their polymerization rates. Concentration of the monomers in the polymerization mixture was increased to monitor conversion of each monomer by 1H NMR spectroscopy. As shown in Fig. 4a, ca. 50% of double bonds in DVB was consumed after 80 h of polymerization while 23% and 3% of styrenic and maleimide double bonds in BMI remained in the polymerization mixture only after 4 h, respectively. The polymerization mixture containing S and BMI became gelled after 5 h. Given that alternating copolymerization consumes styrenic and maleimide double bonds at identical rates, it appears that pendent maleimide bonds attached to the core might be NMR-silent due to low mobility resulting in discrepancy between the consumption rates of styrenic and maleimide double bonds. Assuming first-order kinetics, we estimated that the consumption rate of double bonds by copolymerization of S and BMI is 43 times higher than DVB polymerization (Fig. 4b).
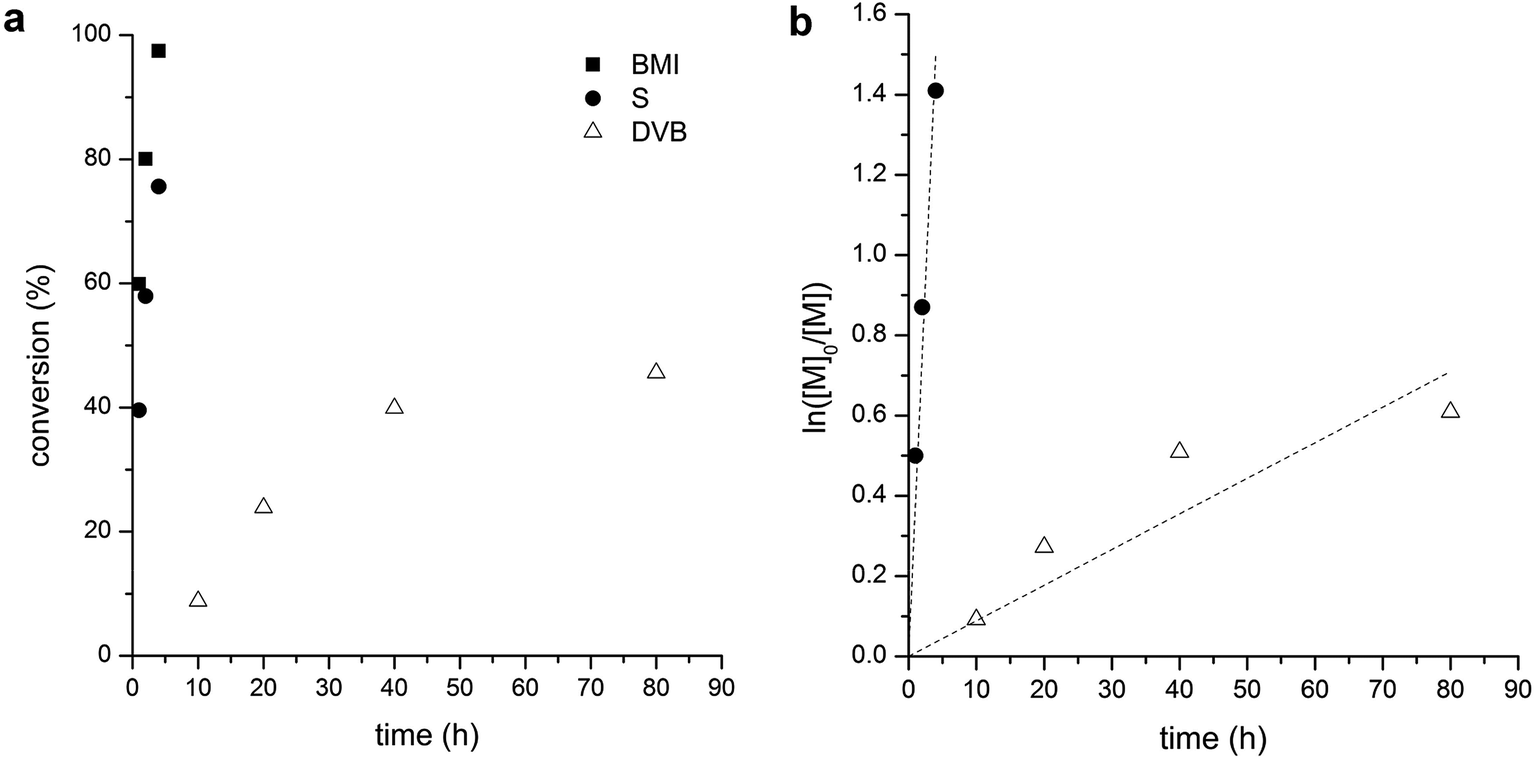 | ||
| Fig. 4 Kinetics of S/BMI copolymerization and DVB polymerization in the presence of PLA-CTA. (a) Time–conversion plot. (b) First-order kinetic plot. Consumption of the double bond of each monomer was estimated by 1H NMR spectroscopy and represented as conversion. In case of S/BMI copolymerization, the dashed line was shown with respect to S concentration. | ||
Size and stability of PLAnPSn (entry 3 in Table 1; Mn,SEC = 458 kg mol−1, Đ = 1.34, fPS = 0.39) in solutions was analyzed by dynamic light scattering (DLS) in comparison with linear PLA-b-PS (Mn,SEC = 55 kg mol−1, Đ = 1.07, fPS = 0.27). While the linear PLA-b-PS was fully soluble in toluene and did not produce strong scattering intensity (Fig. 5a), the heteroarm CCS PLAnPSn existed as stable nanoparticles in toluene because of the cross-linked P(S-alt-BMI) core (Fig. 5b). Hydrodynamic radius (Rh) of the particles was estimated to be 19 nm with polydispersity of 0.40 by Cumulant analysis of the autocorrelation function.
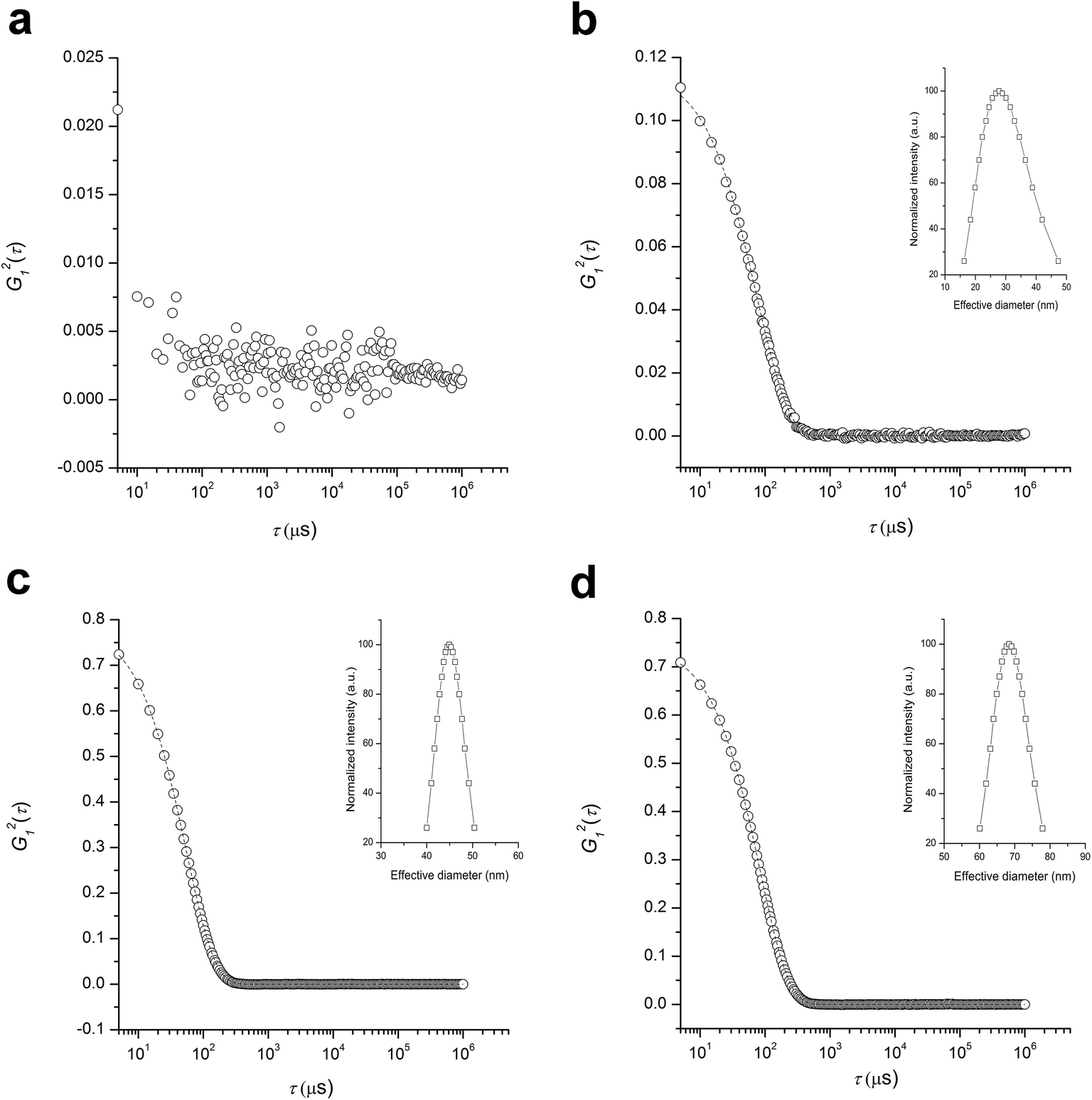 | ||
| Fig. 5 Autocorrelation functions of linear PLA-b-PS (a, c) and heteroarm CCS PLAnPSn (b, d) in (a, b) toluene and (c, d) acetonitrile. Concentration of the solutions was 1 mg mL−1. The insets indicate lognormal distribution of the particle size. | ||
In acetonitrile where PLA is soluble but PS is not, both the linear PLA-b-PS and heteroarm CCS PLAnPSn formed micelles (Fig. 5c and d). While Rh of PLA-b-PS was determined to be 24 nm by Cumulant analysis, Rh of PLAnPSn was 37 nm with polydispersity of 0.051 indicating formation of uniform but larger particles. As Rh of PLAnPSn in acetonitrile was approximately two times larger than Rh in toluene, we hypothesized that PLAnPSn induced intramolecular segregation between PLA and PS arms in acetonitrile and behaved as Janus particles, resulting in formation of supermicelles to minimize exposure of PS arms to acetonitrile.
We characterized the morphology of PLAnPSn in toluene and in acetonitrile by transmission electron microscopy (TEM) (Fig. 6). Samples were prepared by dropping the solution on the TEM grid, evaporating the solvent, and then staining with RuO4. As shown in Fig. 6a, a polymer thin film was fabricated from the toluene solution and microphase separation of PLA (grey) and PS (dark) was evident. This suggests that PLA and PS arms in PLAnPSn can be segregated and further self-assembled to form microphase separated structures, indicating the Janus nature of PLAnPSn. Preliminary small-angle X-ray scattering (SAXS) data of the films cast from toluene solutions supports microphase separation of PLAnPSn (Fig. S7†). Identification of the microphase separated morphologies of PLAnPSn and investigation of the effect of the heteroarm CCS architecture on the microphase separation is in progress. Supermicelles composed of PLAnPSn were also visualized by TEM, revealing the PS micellar core with the diameter of 33 ± 3 nm (Fig. 6b). The TEM image supports our interpretation of the DLS data.
 | ||
| Fig. 6 TEM micrographs of heteroarm CCS PLAnPSn in (a) toluene and (b) acetonitrile. The images were obtained after staining with RuO4. | ||
In summary, we demonstrated heteroarm CCS PLAnPSn can be readily prepared by RAFT copolymerization of BMI with an excess S in the presence of PLA-CTA, exploiting the alternating tendency of maleimide and styrenic double bonds. Investigation of the polymerization kinetics proved formation of the heteroarm CCS PLAnPSn via the “in–out” mechanism, which was further supported by formation of PLAn by stoichiometric copolymerization of S and BMI followed by chain extension with excess S. Compared with DVB that was much less effective for formation of cross-linked core under the identical condition, copolymerization of BMI and S proceeded much faster and efficiently produced CCS polymers demonstrating advantages of employing alternating copolymerization in the CCS polymer synthesis. While the DLS data confirmed the particulate nature of the heteroarm CCS PLAnPSn, TEM and SAXS study indicated that PLA and PS arms in the heteroarm CCS PLAnPSn can undergo intramolecular segregation suggesting utility of the heteroarm CCS polymers as surface active agents and nanostructured materials.
Experimental section
Materials
D,L-Lactide was kindly provided by Purac (Amsterdam, Netherlands), and stored under nitrogen after recrystallization from toluene. 1,8-Diaza-bicyclo[5.4.0]undec-7-ene (DBU) was purchased from Sigma-Aldrich (St. Louis, MO) and also stored under nitrogen. Benzoic acid (99.5%) was purchased from Duksan (Daejeon, Korea) and used as received. Styrene (S, 99%) and divinylbenzene (DVB, 80% (tech.)) were purchased from Sigma-Aldrich and purified by passing through a basic alumina column prior to polymerization. 1,2-Bis(maleimidoethane) (BMI, 98%) was purchased from TCI (Tokyo, Japan) and used as received, or synthesized following a literature procedure.37 2-(Dodecylthiocarbonothioylthio)-2-methylpropionic acid (CTA) was also prepared according to a literature procedure.38 Azobisisobutyronitrile (AIBN, 98%) was purchased from Junsei (Tokyo, Japan) and purified by recrystallization in methanol. HPLC grade dichloromethane (DCM) and N,N-dimethylformamide (DMF) were purchased from Daejung (Siheung, Korea) and J. T. baker (Center Valley, PA), respectively. They were purified using a solvent purification system (C&T International, Suwon, Korea) and used as polymerization solvents. For light scattering experiments, HPLC grade toluene and anhydrous acetonitrile (99.8%) were used which were purchased from Burdick & Jackson (Morristown, NJ) and Sigma-Aldrich, respectively. Toluene was further purified with the solvent purification system prior to use.Methods
1H nuclear magnetic resonance (NMR) spectroscopy was conducted using Bruker Avance 300 and 400 MHz (Billerica, MA) spectrometers using the residual NMR solvent signal as an internal reference. Size exclusion chromatography (SEC) was performed in chloroform at 35 °C on an Agilent 1260 Infinity system (Santa Clara, CA) equipped with a refractive index (RI) detector and three PLgel 10 μm Mixed-B columns in series with a molar mass range 500–10000000 g mol−1. The number average molar masses (Mn,SEC) and dispersities (Đ) of the polymers were calculated relative to linear polystyrene (PS) standards (EasiCal) purchased from Agilent Technologies. Dynamic light scattering (DLS) measurements were performed on a Brookhaven 90Plus/BI-MAS particle size analyzer (Holtsville, NY) at wavelength of 658 nm with scattering angle of 90°. Samples were prepared at a concentration of 1 mg mL−1 and filtered through 0.2 μm PTFE syringe filters prior to the measurements. Transmission electron microscopy (TEM) was performed on a Jeol JEM-2100F field-emission transmission electron microscope (Tokyo, Japan) with acceleration voltage of 200 kV. Samples were prepared on 300 mesh carbon-coated copper grids by dropping the solution (1 mg mL−1) and evaporation of the solvent. The samples were stained by exposing RuO4 vapor prior to imaging. Synchrotron small-angle X-ray scattering (SAXS) experiments were performed at 9A beam line in the Pohang Accelerator Laboratory (PAL). Free-standing polymer films were prepared by casting toluene solutions of PLAnPSn shown in Table 1 and drying under vacuum, and subjected to SAXS experiments at rt. A monochromatized X-ray radiation source of 11.54 keV (0.1074 nm) with the sample-to-detector distance of 6.548 m was used. Scattering intensity was monitored by a Mar 165 mm diameter CCD detector with 2048 × 2048 pixels. The two-dimensional scattering patterns were azimuthally integrated to afford one-dimensional profiles presented as scattering vector (q) versus scattered intensity, where the magnitude of scattering vector is given by q = (4π/λ)sinθ.
Weight-average absolute molecular masses (Mw,MALLS) were determined by using a multi-angle laser light scattering (MALLS) detector attached to SEC. A Thermo Spectra System P1000 SEC instrument (Waltham, MA) equipped with Shodex (Tokyo, Japan) KF-805, KF-802.5 columns in series and a Wyatt DAWN8+ MALLS detector (Santa Barbara, CA) was used with tetrahydrofuran (THF) as eluent at 40 °C. dn/dc was estimated based on the RI intensity of the sample divided by the injected mass. The average number of the polymer arm per star polymer (n) was calculated from Mw,MALLS by the following equation assuming nPLA = nPS and the core is exclusively composed of P(S-alt-BMI):
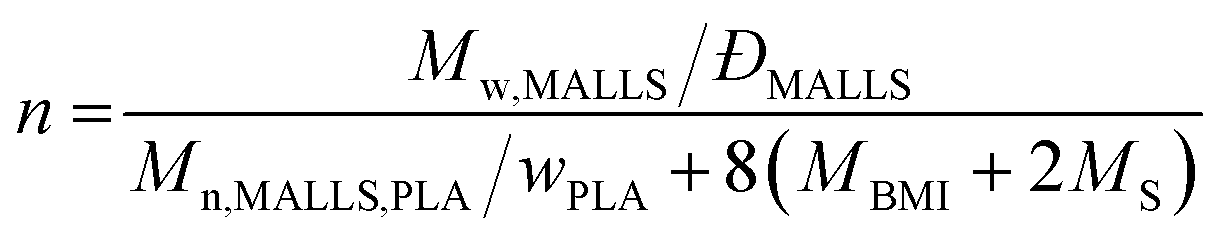
Weight fraction of PLA (wPLA) was determined by 1H NMR spectroscopy. Mn,MALLS,PLA was estimated from the MALLS-SEC analysis of PLA-CTA, which gave Mw,MALLS = 22 kg mol−1 with ĐMALLS = 1.02. MBMI and MS represent molar masses of BMI and S, respectively.
Copolymerization kinetics of S (excess) and BMI in the presence of PLA-CTA
A polymerization mixture containing S (0.5 mL, 4.36 mmol), BMI (0.0154 g, 0.0696 mmol), PLA-CTA (Mn,NMR = 30 kg mol−1, 0.268 g, 8.7 μmol), AIBN (0.143 mg, 0.87 μmol, added as 1% (w/w) solution in benzene), and DMF (1 mL) was prepared and transferred into an ampoule. After three cycles of freeze–pump–thaw, the ampoule was flame-sealed under vacuum and then immersed in an oil bath preset to 60 °C. After stirring for 36 h, the ampoule was cooled to rt and then opened to stop the polymerization. An aliquot was taken from the polymerization mixture to estimate conversion by 1H NMR analysis, which was 25% in this case. The polymerization mixture was diluted with DCM and precipitated in methanol. The polymer was obtained by filtration and dried under vacuum at rt overnight (0.31 g, 74% yield). The rest of the polymer was recovered by evaporation of the filtrate and gave an indistinguishable SEC trace compared with the precipitated one. 1H NMR analysis of the polymer indicated the polymer contained 70% of PLA and 30% of PS (see Fig. S3†). SEC analysis of the polymer gave Mn,SEC = 487 kg mol−1 and Đ = 1.37.As a comparison, RAFT polymerization of S without BMI in the presence of PLA-CTA was conducted following the identical procedure and condition described above. The resulting PLA-b-PS was composed of 73% of PLA and 27% of PS, and the molecular weight was estimated as Mn,SEC = 55 kg mol−1 and Đ = 1.07 by SEC analysis (see Fig. S4†). Using the identical composition of the polymerization mixture ([S]:[BMI]:[PLA-CTA] = 500:8:1), the copolymerization kinetics was investigated and summarized in Table 1. We also investigated the effect of [S]:[BMI]:[PLA-CTA] ratios on the polymerization fixing the polymerization time as 36 h, and summarized the results in Table S1.†
Stoichiometric copolymerization of S and BMI in the presence of PLA-CTA and subsequent chain extension with S
A polymerization mixture containing S (0.0165 mL, 16 mmol), BMI (0.0153 g, 8 mmol), PLA-CTA (Mn,NMR = 22 kg mol−1, 0.2 g, 8.7 μmol), AIBN (0.143 mg, 0.87 μmol, added as 1% (w/w) solution in benzene), and DMF (1 mL) was prepared and transferred into an ampoule. After three cycles of freeze–pump–thaw, the ampoule was flame-sealed under vacuum and then immersed in an oil bath preset to 60 °C. After stirring for 24 h, the ampoule was cooled to rt and then opened. The polymerization mixture was precipitated in methanol. PLAn was obtained by filtration and dried under vacuum at rt overnight (0.16 g, 67% yield). A SEC trace of PLAn is shown in Fig. 2a (solid line).PLAn was further used as a macro-CTA for chain extension with S. A polymerization mixture containing PLAn (0.032 g), S (0.124 g, 1.19 mmol), AIBN (0.143 mg, 0.87 μmol), and DMF (1 mL) was prepared and transferred into an ampoule. After three cycles of freeze–pump–thaw, the ampoule was flame-sealed under vacuum and then immersed in an oil bath preset to 60 °C. After stirring for 12 h, the ampoule was cooled to rt and then opened. The polymerization mixture was precipitated in methanol and PLAnPSn was obtained by filtration and dried under vacuum at rt overnight. A SEC analysis of PLAnPSn is shown in Fig. 2a (dashed line).
Stoichiometric copolymerization of S and BMI in the presence of PLA-CTA followed by addition of S
A polymerization mixture containing S (0.015 mL, 0.14 mmol), BMI (0.0154 g, 0.07 mmol), PLA-CTA (Mn,NMR = 30 kg mol−1, 0.268 g, 8.7 μmol), AIBN (0.143 mg, 0.87 μmol, added as 1% (w/w) solution in benzene), and DMF (1 mL) was prepared and transferred into an Schlenk flask. After three cycles of freeze–pump–thaw, the flask was filled with Ar gas and then immersed in an oil bath preset to 60 °C. After stirring for 24 h, degassed S (0.485 mL, 4.22 mmol) was added to the flask and the polymerization mixture was further stirred for 12 h. After cooling to rt, the polymerization mixture was diluted with DCM and precipitated in methanol. PLAnPSn was obtained by filtration and dried under vacuum at rt overnight. A SEC trace of the polymer is shown in Fig. 2b.Polymerization kinetics of S/BMI and DVB in the presence of PLA-CTA
A polymerization mixture containing S (0.165 mL, 160 mmol), BMI (0.158 g, 80 mmol), PLA-CTA (Mn,NMR = 22 kg mol−1, 0.200 g, 9.0 μmol), AIBN (0.043 g, 2.6 μmol, added as 1% (w/w) in benzene), and DMF (5 mL) was prepared and transferred into an ampoule. 4 ampoules containing the identical polymerization mixture were prepared. Each ampoule was flame-sealed after three cycles of freeze–pump–thaw and then immersed in an oil bath preset to 60 °C. After stirring for 1, 2, 4, and 6 h, each ampoule was taken out of the bath and opened. Conversion was estimated by 1H NMR analysis of the aliquot.In case of DVB kinetics, DVB (0.15 mL, 120 mmol) was used instead of S and BMI following the procedure described above. Each ampoule was quenched after stirring for 10, 20, 40, and 80 h at 60 °C, respectively.
Acknowledgements
This research was supported by the HRHR Project funded by KAIST. The authors thank Dr Byoung Gak Kim at Korea Research Institute of Chemical Technology and Insung Chromatech for the use of MALLS-SEC. Experiments at Pohang Accelerator Laboratory (PAL) were supported in part by Ministry of Science, ICT and Future Planning of Korea and POSTECH. TEM imaging was conducted in Korea National NanoFab Center supported by Nano Material Technology Development Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (2009-0082580).References
- N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 857–871 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068–1132 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS.
- K. Khanna, S. Varshney and A. Kakkar, Polym. Chem., 2010, 1, 1171–1185 RSC.
- G. Floudas, S. Pispas, N. Hadjichristidis, T. Pakula and I. Erukhimovich, Macromolecules, 1996, 29, 4142–4154 CrossRef CAS.
- S. Sioula, Y. Tselikas and N. Hadjichristidis, Macromol. Symp., 1997, 117, 167–174 CrossRef CAS.
- D. Voulgaris, C. Tsitsilianis, F. J. Esselink and G. Hadziioannou, Polymer, 1998, 39, 6429–6439 CrossRef CAS.
- K. Ishizu and S. Uchida, Prog. Polym. Sci., 1999, 24, 1439–1480 CrossRef CAS.
- V. Grayer, E. E. Dormidontova, G. Hadziioannou and C. Tsitsilianis, Macromolecules, 2000, 33, 6330–6339 CrossRef CAS.
- A. O. Moughton, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2012, 45, 2–19 CrossRef CAS.
- P. G. de Gennes, Angew. Chem., Int. Ed., 1992, 31, 842–845 CrossRef.
- A. Walther and A. H. E. Müller, Chem. Rev., 2013, 113, 5194–5262 CrossRef CAS PubMed.
- Y. Chang, W. C. Chen, Y. J. Sheng, S. Jiang and H. K. Tsao, Macromolecules, 2005, 38, 6201–6209 CrossRef CAS.
- A. Kiriy, G. Gorodyska, S. Minko, M. Stamm and C. Tsitsilianis, Macromolecules, 2003, 36, 8704–8711 CrossRef CAS.
- Z. M. Wu, H. Liang, J. Lu and W. L. Deng, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3323–3330 CrossRef CAS.
- X. Shi, W. Zhou, Q. Qiu and Z. An, Chem. Commun., 2012, 48, 7389–7391 RSC.
- A. Biencow, J. F. Tan, T. K. Goh and G. G. Qiao, Polymer, 2009, 50, 5–32 CrossRef.
- C. Tsitsilianis and D. Voulgaris, Macromol. Chem. Phys., 1997, 198, 997–1007 CrossRef CAS.
- H. Gao, N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2005, 38, 5995–6004 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Macromol. Symp., 2010, 291–292, 12–16 CrossRef CAS.
- For example, see H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 11828–11834 CrossRef CAS PubMed.
- For example, see T. Tsoukatos, S. Pispas and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 320–325 CrossRef CAS.
- G. Zheng and C. Pan, Polymer, 2005, 46, 2802–2810 CrossRef CAS.
- Q. Zheng, G. H. Zheng and C. Y. Pan, Polym. Int., 2006, 55, 1114–1123 CrossRef CAS.
- A. Gregory and M. H. Stenzel, Prog. Polym. Sci., 2012, 37, 38–105 CrossRef CAS.
- G. Zheng and C. Pan, Macromolecules, 2006, 39, 95–102 CrossRef CAS.
- X. Shi, M. Miao and Z. An, Polym. Chem., 2013, 4, 1950–1959 RSC.
- T. G. McKenzie, E. H. H. Wong, Q. Fu, A. Sulistio, D. E. Dunstan and G. G. Qiao, ACS Macro Lett., 2015, 4, 1012–1016 CrossRef CAS.
- D. Benoit, C. J. Hawker, E. E. Huang, Z. Lin and T. P. Russell, Macromolecules, 2000, 33, 1505–1507 CrossRef CAS.
- A. P. Bapat, J. G. Ray, D. A. Savin, E. A. Hoff, D. L. Patton and B. S. Sumerlin, Polym. Chem., 2012, 3, 3112 RSC.
- Q. Liu and Y. Chen, Macromol. Chem. Phys., 2007, 208, 2455–2462 CrossRef CAS.
- A. W. Bosman, A. Heumann, G. Klaerner, D. Benoit, J. M. J Fréchet and C. J. Hawker, J. Am. Chem. Soc., 2001, 123, 6461–6462 CrossRef CAS PubMed.
- R. Erhardt, A. Böker, H. Zettl, H. Kaya, W. Pyckhout-Hintzen, G. Krausch, V. Abetz and A. H. E. Müller, Macromolecules, 2001, 34, 1069–1075 CrossRef CAS.
- M. Seo, D. Moll, C. Silvis, A. Roy, S. Querelle and M. A. Hillmyer, Ind. Eng. Chem. Res., 2014, 53, 18575–18579 CrossRef CAS.
- J. Oh and M. Seo, ACS Macro Lett., 2015, 4, 1244–1248 CrossRef CAS.
- We note that “homopolymerization” of DVB in this study is technically copolymerization of three different monomers as we used tech. grade DVB (80%) which contains m- and p-isomers as well as ethylstyrene.
- (a) D. Habibi and O. Marvi, ARKIVOC, 2006, 13, 8 Search PubMed; (b) L. Wei, L. Cao and Z. Xi, Angew. Chem., Int. Ed., 2013, 52, 6501–6503 CrossRef CAS PubMed.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 and Fig. S1–S7. See DOI: 10.1039/c6ra07527d |
| This journal is © The Royal Society of Chemistry 2016 |