
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel antimicrobial target—expanded and revisited mode of action of pantothenamides†
Aleksandra
Maršavelski
Ruđer Bošković Institute, Computational Organic Chemistry and Biochemistry Group, Bijenička cesta 54, HR-10000, Zagreb, Croatia. E-mail: aleksandra.marsavelski@irb.hr
First published on 3rd May 2016
Abstract
Pantothenamides are analogs of pantothenic acid (vitamin B5), which is a natural precursor of coenzyme A (CoA). It has been shown that these compounds, predominantly N-substituted pantothenamides, possess antimicrobial activity against various pathogenic bacteria such as E. coli and S. aureus. It is widely accepted that these compounds act through combined inhibition of coenzyme A and fatty acid synthesis. However, the precise mechanism of action remains unrevealed. Here is reported the identification of a novel target of pantothenamides, never considered before. Molecular dynamics simulations together with free energy calculations reveal that the hydrophobic pocket of the acyl carrier protein (ACP) binds N-pentylpantothenamide. Consequently, the sequestration of the acyl chain attached to the Ppant prosthetic arm is defunct since the inhibitor occupies the hydrophobic core of the ACP. Thus, the acyl chain remains solvent-exposed and susceptible to hydrolysis. Moreover, the ACP with N-pentylpantothenamide bound could change its chain-flipping ability as well as its interaction propensity towards downstream enzyme partners of the fatty acid synthesis pathway, which could result in the suppression of the fatty acid synthesis rate.
Introduction
The concept of anti-vitamins, inhibitors of metabolic processes that involve a specific vitamin, was established in the early 1930s, with the discovery of the sulfonamides. Since anti-vitamins structurally resemble vitamins, they easily enter bacterial cells through various transport systems. This idea led to the propulsive search for new anti-vitamin compounds, which resulted in the discovery of N-substituted pantothenamides, pantothenic acid analogs. These analogs, in the first place prototypic N-pentylpantothenamide (N5-Pan) and N-heptylpantothenamide (N7-Pan), are bacteriostatic compounds.1 Pantothenamides are also shown to be potent inhibitors of the growth of the human malaria parasite Plasmodium falciparum.2 However, the antimicrobial mode of action of the pantothenate antimetabolites is still under debate. The accepted mechanism is shown in Fig. 1.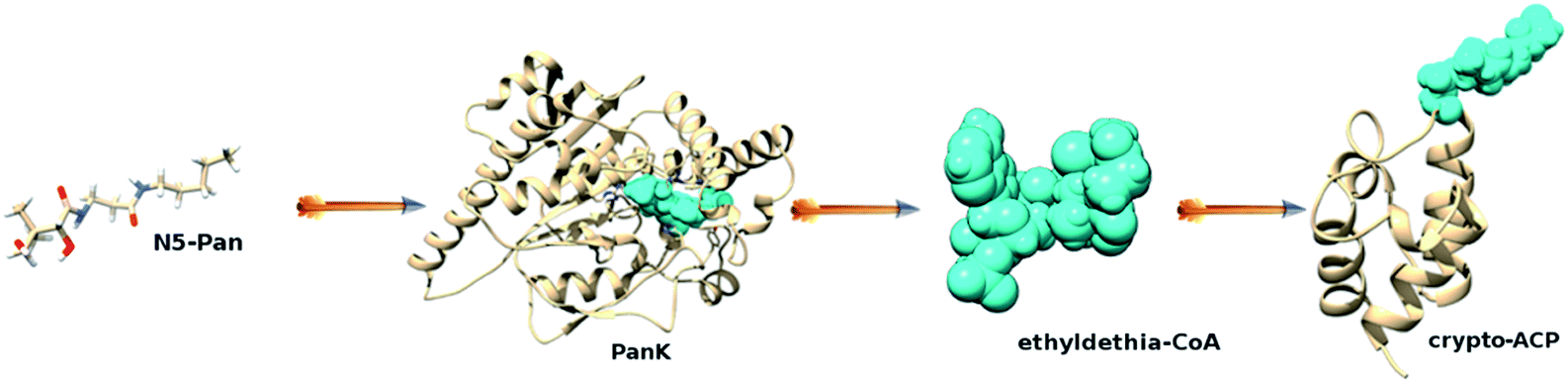 | ||
| Fig. 1 Schematic representation showing the mode of action of N-substituted pantothenamides through combined inhibition of the coenzyme A and fatty acid synthesis. When N5-Pan is present in E. coli, PanK, an enzyme of the coenzyme A (CoA) biosynthetic pathway, utilizes the N5-Pan instead of pantothenate. Resulting compound ethyldethia-CoA then inhibits the CoA-utilizing enzymes or proteins. For instance, acyl carrier protein, a central player in the synthesis of all fatty acids, is post-translationally modified with a moiety derived from ethyldethia-CoA. Such crypto-ACP is incapable of binding intermediates of the fatty acid synthesis pathway, causing the suppression of the fatty acid synthesis. | ||
Pantothenate kinase (PanK), an enzyme of the coenzyme A (CoA) biosynthetic pathway, utilizes N-pentylpantothenamide as a substrate instead of pantothenate. The resulting compound ethyldethia-CoA then inhibits the CoA-utilizing enzymes or proteins.3 For instance, apo acyl carrier protein (apo-ACP), is post-translationally modified with 4′-phosphopantetheine (Ppant) prosthetic group derived from CoA, resulting in a biologically functional holo-ACP. However, in the presence of ethyldethia-CoA, apo-ACP is modified with a dysfunctional prosthetic group (Fig. 1), which leads to the accumulation of dysfunctional ethyldethia-ACP (crypto-ACP).4 Moreover, in S. aureus, next to ACP, pantothenamide antimetabolites are incorporated into Dcp,5 which is a small carrier protein required for the transfer of D-alanine to lipoteichoic acid in Gram-positive bacteria. Such ethyldethia-ACP, deprived of the terminal thiol group, is no longer capable of binding and delivering growing acyl chains to the partner enzymes of the fatty acid biosynthesis pathway. Thus, posttranslational modification of apo-ACP that results in a dysfunctional prosthetic group, incapable of binding various intermediates of the fatty acid synthesis, was seen as a bacteriostatic. Since the gene for the enzyme that hydrolyzes holo-ACP to apo-ACP was unknown and, therefore, could not be expressed, it was hypothesized that ACP phosphodiesterase was unable to hydrolyze ethyldethia-ACP, a defective ACP. Consequently, it was concluded that the ethyldethia-ACP accumulation leads to bacterial cell death. A few years later, Thomas and Cronan have shown that ACP phosphodiesterase is capable of hydrolyzing ethyldethia-ACP both in vivo and in vitro, which ruled out the accumulation of the ethyldethia-ACP as a cause of bacterial cell death.6 Moreover, it was shown that in the presence of N5-Pan neither overexpression of enzymes of the coenzyme A synthesis pathway (PanK, CoaD, CoaE, and Dfp)6 nor supplementation of the growth medium with pantothenate4 overcome the growth inhibition. These results questioned the inhibition of CoA synthesis, as a possible mechanism of action of pantothenamides. Nevertheless, it was concluded that the mode of action of pantothenamides is probably through the combined inhibition of the coenzyme A and fatty acid synthesis (Fig. 1), leaving the precise mechanism unexplained.
Puzzled by ambiguous experimental data and intrigued by the structural similarities between (a) N-substituted pantothenamides and (b) saturated acyl chain covalently linked to 4′-phosphopantetheine (Ppant) prosthetic group, an alternative cellular target for the pantothenamides is considered. In the study reported here, the author was interested in whether the hydrophobic core of the acyl carrier protein could be a target for N-substituted pantothenamides. Moreover, if this proves to be true, what are the potential consequences of the binding? Classical unbiased molecular dynamics simulations together with the binding free energy calculations of N-pentylpantothenamide bound inside the hydrophobic core of various forms of E. coli acyl carrier proteins were performed to address these questions.
Materials and methods
Model preparation
Acyl carrier protein takes a central part in the fatty acid synthesis. Fatty acids are synthesized in the repetitive cycle consisting of the following reactions: condensation, reduction, dehydration, and reduction. All of the intermediates are products of each of these reaction steps, and all of them are attached to the Ppant prosthetic group of the holo-ACP that serves as a carrier of a growing acyl chain. Thus, ACP operates by exposing and shielding its cargo from the solvent and non-partner enzyme for one particular step in the synthesis. Substrate delivery demands the first three carbon atoms of the acyl intermediate to be exposed from the hydrophobic pocket of the ACP in order to reach the catalytic residues of the corresponding FAS enzyme. The reactions are repeated until, for instance, the palmitoyl product (fully saturated C16 product) is synthesized. NMR structural analysis7 revealed that ACP adopts a unique conformation for each intermediate attached. Some of these intermediates are 3-oxoacyl-[acyl-carrier-protein], 3-hydroxyacyl-[acyl-carrier-protein], 2-trans-enoyl-[acyl-carrier-protein], and fully saturated acyl-ACP, and all of them could be placed in the hydrophobic pocket and exposed to the corresponding partner enzyme when necessary. Up to the present time, molecular dynamics simulations are published for apo-, holo-, and saturated acyl-forms of E. coli ACP,8 and for intermediates in the fatty acid synthesis, 3-oxoacyl-[acyl-carrier-protein], 3-hydroxyacyl-[acyl-carrier-protein] and 2-trans-enoyl-[acyl-carrier-protein] of E. coli.9 Moreover, all of them captured the transition from the solvent-exposed conformation to the solvent shielded, which is, to date, well-known fact, studied either experimentally or computationally. However, all of them failed to show reverse motion, the extrusion from the hydrophobic pocket of the ACP. The reason lies in the fact that the exposure of the acyl chain would never happen per se. In other words, acyl carrier protein will never expose, by itself, acyl chain attached to its prosthetic group to a solvent. To do so, ACP requires a suitable protein partner with whom it can accomplish appropriate protein–protein contacts to fulfill its biological purpose, which is exposing and subsequent delivery of the acyl chain to the active site of the partner enzyme.In this regard, eight protein structures of the acyl carrier protein are modeled (Table 1).
| Model | Simulation time per model | Number of independent simulations |
|---|---|---|
| Apo-ACP | 600 ns | 1 |
| Apo-ACP + N5-Pan | 600 ns | 3 |
| Holo-ACP | 600 ns | 1 |
| Holo-ACP + N5-Pan | 600 ns | 3 |
| Hexanoyl-ACP | 600 ns | 1 |
| Hexanoyl-ACP + N5-Pan | 600 ns | 3 |
| Decanoyl-ACP | 600 ns | 1 |
| Decanoyl-ACP + N5-Pan | 600 ns | 3 |
Starting structures of apo-ACP, an inactive acyl carrier protein without the prosthetic group, and apo-ACP with N5-Pan bound in the pocket are shown in Fig. 2A and B, respectively. Starting structures of holo-ACP, hexanoyl-ACP, and decanoyl-ACP with the solvent-exposed prosthetic arm as well as acyl chains attached to it are shown in Fig. 2C, E and G. Holo-ACP + N5-Pan, hexanoyl-ACP + N5-Pan, and decanoyl-ACP + N5-Pan are ACPs with the solvent-exposed prosthetic groups with simultaneously bound N5-Pan in the hydrophobic core of the ACPs (Fig. 2D, F and H). Apo-ACP was modeled using the X-ray structure of E. coli apo-ACP10 (ID 1T8K, 1.10 Å). The crystal structure of E. coli hexanoyl-ACP11 (ID 2FAC, 1.76 Å) was used to model manually N5-Pan accommodated in the hydrophobic core from the buried hexanoyl chain. This structure was later used to model apo-ACP + N5-Pan, holo-ACP + N5-Pan, hexanoyl-ACP + N5-Pan, and decanoyl-ACP + N5-Pan, by overlapping empty ACPs with ACP occupied with the N5-Pan and transferring the N5-Pan coordinates. ACPs with the solvent-exposed prosthetic group (Ppant), and both hexanoyl- and decanoyl-chains linked to the prosthetic group, were modeled based on the crystal structure of the crosslinked E. coli ACP–FabA complex12 (4KEH, 1.9 Å). The coordinates of FabA enzyme from the 4KEH crystal structure were deleted, while the coordinates of the ACP with 4′-phosphopantetheine group with the sulphonyl-3-alkyne-based probe linked to it, which resembles decanoyl moiety, were retained. The cross-linking probe was modified manually to decanoyl- and hexanoyl-acyl moieties. By deleting sulphonyl-3-alkyne-based probe, the solvent exposed 4′-phosphopantetheine (Ppant) was modeled. In this way, the prosthetic group as well as acyl moieties pointed vertically up from the ACP were obtained, thus imitating situation when the Ppant + acyl chain is bound to one of the bacterial fatty acid synthase II enzymes.
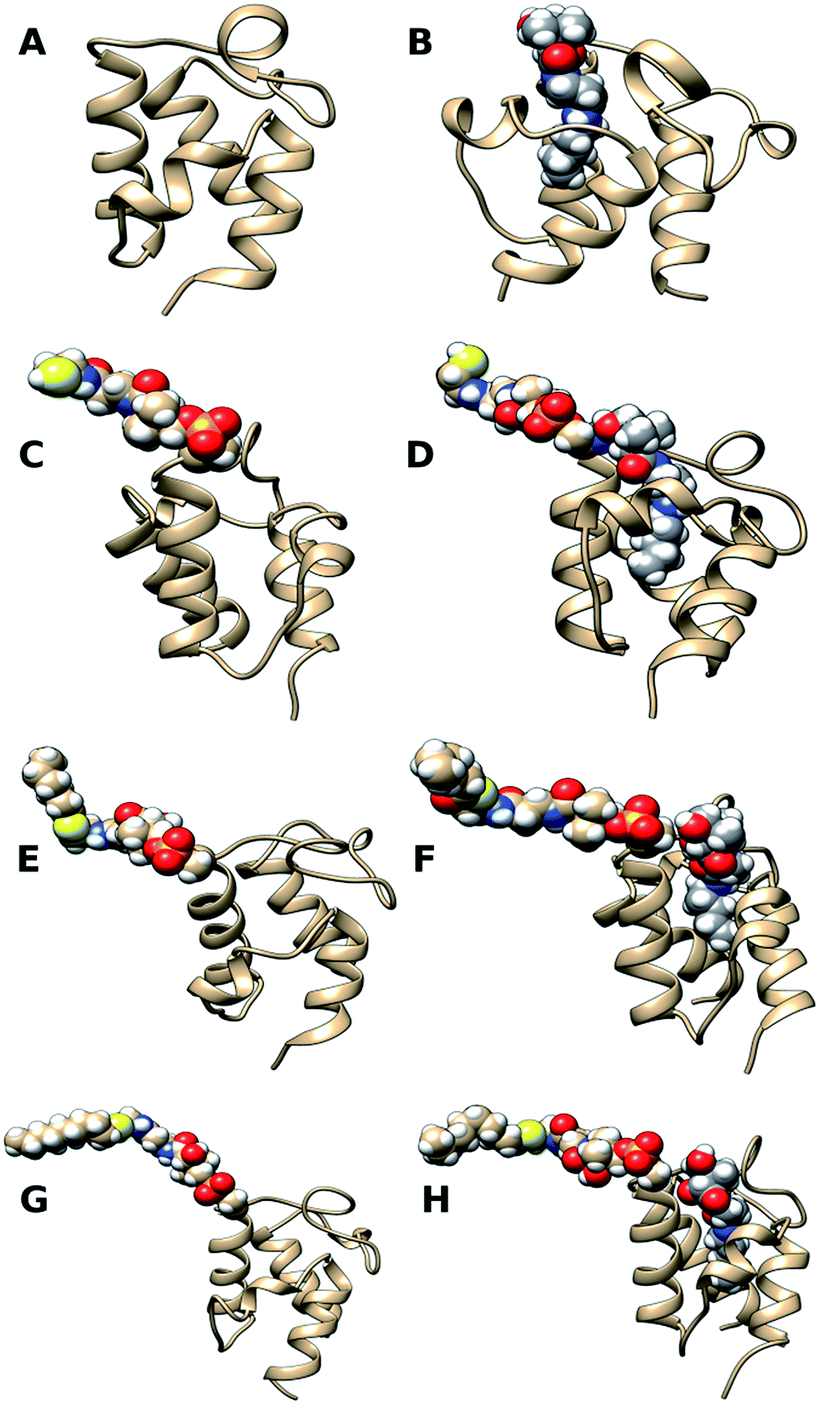 | ||
| Fig. 2 Starting structures for MD simulations. (A) Apo-ACP, acyl carrier protein lacking its prosthetic group. (B) Apo-ACP in complex with N5-Pan. (C) Holo-ACP, acyl carrier protein with the solvent-exposed prosthetic group. (D) Holo-ACP, acyl carrier protein with the solvent-exposed prosthetic group, and simultaneously bound N5-Pan. (E) Hexanoyl-ACP, acyl carrier protein with the solvent-exposed hexanoyl acyl chain linked to the prosthetic group. (F) Hexanoyl-ACP, acyl carrier protein with the solvent-exposed hexanoyl acyl chain linked to the prosthetic group, and simultaneously bound N5-Pan in the hydrophobic pocket of the ACP. (G) Decanoyl-ACP, acyl carrier protein with the solvent-exposed decanoyl acyl chain linked to the prosthetic group. (H) Decanoyl-ACP, acyl carrier protein with the solvent-exposed decanoyl acyl chain linked to the prosthetic group, and simultaneously bound N5-Pan in the hydrophobic pocket of the ACP. | ||
Molecular dynamics simulation
All MD simulations were performed with the AMBER ff12SB force field and the general AMBER force field GAFF.13 Partial charges for nonstandard residues (4′-phosphopantetheine, decanoyl-, hexanoyl-acyl moieties, and N5-Pan) were derived using the RESP approach14 with charges calculated based on an RESP fit to an HF/6-31G* electrostatic potential. All protein structures were solvated in a truncated octahedral box of TIP3P water,15 such that no solute atom was less than 10 Å from box edges. Na+ ions were added for neutralization. The monovalent ion parameters16 for TIP3P water implemented in AMBER were used. The resulting solvated system was energy-minimized in 3 cycles. In the first cycle, proteins were fixed using a restraint on each protein atom while water, ions, and N5-Pan were allowed to move using 500 steps of steepest descent minimization, followed by 1000 steps of conjugate gradient minimization. In the second cycle, proteins were fixed using a restraint on backbone atoms only while side chains of the protein, water, ions and N5-Pan were allowed to move using 500 steps of steepest descent minimization, followed by 2000 steps of conjugate gradient minimization. In the last cycle, the whole system was energy-minimized for 5000 steps, including 1500 steps of steepest descent minimization and 3500 steps of conjugate gradient minimization. Optimized systems were gradually heated from 0 to 300 K and equilibrated during 50 ps using NVT conditions, and subjected to productive, unconstrained MD simulations at constant pressure (1 atm) and temperature (300 K) using the Berendsen barostat17 and Langevin thermostat,18 respectively. The production runs, each 600 ns in length, were performed on the GPU (NVIDIA Tesla M2075) using the pmemd.CUDA19–21 engine of AMBER12.22 Bonds involving hydrogen atoms were constrained using the SHAKE algorithm.23 The time step was 2 fs. The Particle Mesh Ewald method24 was applied to calculate long-range electrostatic interactions. Nonbonded interactions were truncated at 10.0 Å. For all protein complexes, three independent MD simulations starting from the same initial structure, but with different random number seeds for initial velocity assignment were performed. Therefore, three independent 600 ns MD simulations for each complex were run, giving a sum of 1.8 μs of total MD simulations for each complex and a total of 7.2 μs across all four complexes. In this way, by performing extensive MD simulations in triplicates for each complex, the author wanted to check the convergence of the simulations as well as the relevance and reliability of the simulations and calculated binding free energies. This approach is chosen because it provides the more efficient sampling of the conformational space since the complexes of ACP have been manually prepared, where a possibility of imposed stability is higher.Free energy calculations
The binding free energy of N5-Pan in apo-ACP + N5-Pan, holo-ACP + N5-Pan, hexanoyl-ACP + N5-Pan and deacnoyl-ACP + N5-Pan complexes, ΔGbind, was calculated according to the following equation:| ΔGbind = 〈Gcomplex〉 − 〈Gacyl carrier protein〉 − 〈GN5-PAN〉 | (1) |
| Gcomplex/acyl carrier protein/N5-PAN = EMM + Gsolv − TΔSMM | (2) |
| Gnonpolar = γSASA + b | (3) |
Data analysis
All MD trajectories were analyzed using the CPPTRAJ29 available in the AmberTools15. Cluster analysis was performed using DBscan30 clustering method where the coordinate RMSD was used as the distance metric. DBscan clustering was performed on a subset of the protein backbone atoms. A distance cutoff ε of 0.9 Å between clusters was used and a minimum of 25 points was required to form a cluster. VMD,31 Pymol32 software as well as Chimera,33 were employed for the protein and trajectory visualization, where the last was used for making protein movies. Graphs were plotted using Xmgrace34 and Gnuplot.35 The GNU Image Manipulation Program (GIMP, http://www.gimp.org/) was used for figure preparation.Results and discussion
Root-mean-square deviations (RMSD) of the uncomplexed ACPs and ACPs in complex with N5-Pan throughout the simulation period showed no significant changes in the backbone of the proteins, implying that the binding of N5-Pan in the hydrophobic core of the ACP does not affect the protein stability (Fig. S1†). All simulated ACPs complexes retained the N5-Pan in the hydrophobic core of the ACP during the simulated period, except for decanoyl-ACP + N5-Pan complexes, where the decanoyl chain has pushed out the N5-Pan from the pocket in two simulations out of three, while in the third simulation, N5-Pan stayed bound during the entire simulation period. The holo-ACP + N5-Pan complexes were stable during the simulated period; i.e. N5-Pan remained bound in the hydrophobic core of the ACPs (Fig. S2†). The same behavior is observed for the apo-ACP + N5-Pan complexes (Fig. S3†). Moreover, both acyl-bearing ACPs (hexanoyl- and decanoyl-ACP) exhibit the transition from the solvent-exposed to the solvent-shielded acyl chains, which is an inherent feature of class II acyl-bearing ACPs (Fig. S4 and S5, and Movies S1 and S2†). On the other hand, the hexanoyl-ACP + N5-Pan complexes did not show the transition of hexanoyl chain in the hydrophobic core (Fig. 3 and S6, and Movie S3†). Interestingly, in two simulations out of three, decanoyl-ACP + N5-Pan did accommodate decanoyl chain in the hydrophobic core while N5-Pan has been simultaneously pushed out (Fig. 3, S7, and Movie S4†), while in the third simulation of the decanoyl-ACP + N5-Pan complex, N5-Pan stayed bound during the entire simulation period, i.e. ACP has not accommodated its decanoyl chain in the hydrophobic pocket (Movie S5†). | ||
| Fig. 3 Hexanoyl-ACP did not sequester its acyl chain in the hydrophobic pocket occupied with N5-Pan shown in cyan (left); decanoyl-ACP did sequester its acyl chain in the hydrophobic pocket while N5-Pan has been simultaneously pushed out (right). | ||
All three simulations of the hexanoyl-ACP in complex with N5-Pan failed to show the transition from the solvent-exposed acyl chain to the solvent-shielded conformation. It seems that hexanoyl chain could not compete in terms of hydrophobicity with N5-Pan thus failed to enter into the hydrophobic core (Movie S3†), which is not the case with decanoyl chain whose hydrophobic character prevails over that of the N5-Pan, which has been pushed out of the hydrophobic core in two out of three simulations (Movie S4†).
N5-Pan binding to the acyl carrier protein did not influence the overall fold consisting of four α helices. Secondary structure elements of ACPs are well preserved upon inhibitor binding during all MD simulations (Fig. S8†). The binding free energies were calculated for all complexes where N5-Pan stayed bound during the entire simulation period of 600 ns. The calculated binding free energies across 100 frames shown in Table 2 and 250 frames (Table S1†) indicate that the N5-Pan binding in the hydrophobic pockets of apo-, holo-, hexanoyl- and decanoyl-ACPs is energetically favorable. Estimated binding free energies cluster around a value of −5 kcal mol−1.
| System | VDW | EEL | EPB | ENPOLAR | ΔHgas | ΔHsolv | ΔHbind | TΔS | ΔGbind |
|---|---|---|---|---|---|---|---|---|---|
| a VDW = van der Waals contribution from MM; EEL = electrostatic energy as calculated by the MM force field; EPB = the electrostatic contribution to the solvation free energy calculated by PB; ENPOLAR = nonpolar contribution to the solvation free energy calculated by an empirical model. | |||||||||
| Apo-ACP | −32.1 | −27.3 | 36.7 | −4.6 | −59.4 | 32.1 | −27.3 (±3.3) | −21.4 (±3.4) | −5.9 |
| Apo-ACP | −33.4 | −27.4 | 40.0 | −4.8 | −60.8 | 35.2 | −25.6 (±4.1) | −20.9 (±4.3) | −4.7 |
| Apo-ACP | −29.7 | −35.4 | 43.5 | −4.7 | −65.1 | 38.8 | −26.3 (±2.8) | −21.0 (±4.8) | −5.3 |
| Holo-ACP | −32.1 | −30.2 | 40.5 | −4.6 | −62.3 | 35.9 | −26.4 (±4.4) | −20.9 (±5.2) | −5.5 |
| Holo-ACP | −37.4 | −21.1 | 38.8 | −4.9 | −58.5 | 33.9 | −24.6 (±4.5) | −19.4 (±4.9) | −5.2 |
| Holo-ACP | −30.2 | −35.2 | 44.1 | −4.5 | −65.4 | 39.6 | −25.9 (±3.8) | −20.1 (±5.1) | −5.8 |
| Hexanoyl-ACP | −32.4 | −36.5 | 46.4 | −4.7 | −68.9 | 41.7 | −27.2 (±4.1) | −22.8 (±3.4) | −4.4 |
| Hexanoyl-ACP | −37.2 | −34.4 | 47.4 | −4.7 | −71.6 | 42.7 | −28.9 (±3.4) | −23.4 (±3.7) | −5.5 |
| Hexanoyl-ACP | −32.0 | −28.5 | 39.0 | −4.6 | −60.5 | 34.4 | −26.1 (±4.0) | −21.6 (±3.7) | −4.5 |
| Decanoyl-ACP | −36.7 | −20.8 | 33.2 | −4.6 | −57.5 | 28.6 | −28.9 (±3.7) | −23.1 (±3.2) | −5.8 |
The most dominant contribution comes from the nonpolar part (vdW + ENPOLAR), which prevails over the combined electrostatic contribution (EEL + EPB). This favorable nonpolar contribution can be rationalized as depicted in Fig. 4, which is the representative structure of the most populated cluster derived from the apoACP + N5-Pan trajectory. Clustering was performed independently for each apoACP + N5-Pan complex, as described in Data analysis section, giving a total of three representative structures from the most populated clusters derived from three trajectories. The three structures show a considerable overlap; the backbone RMSD values between the first and second, and the first and third representative structures from the most populated clusters are 1.10 and 1.01 Å, respectively.
 | ||
| Fig. 4 Hydrophobic pocket of the ACP with N5-Pan bound showing favorable hydrophobic interactions between the pentyl moiety of the N5-Pan and amino acids of the hydrophobic pocket. | ||
The pentyl moiety of the N5-Pan makes hydrophobic contacts with the aromatic and nonpolar amino acids of the hydrophobic pocket of the ACP, namely Phe28, Val29, Leu42, Val43, Leu46, Ile54, Ala59, Ile62, Ala68. Next to favourable hydrophobic contacts formed between pentyl moiety of the N5-Pan and amino acids of the hydrophobic pocket, N-pentylpantothenamide makes hydrogen bonds occasionally with the side-chain hydroxyl group of Thr39, side-chain carboxyl group of Glu60 (Fig. 4) and carbonyl oxygens of the peptide bond of Ala59, Glu60 and Ile62 (Tables S2–S5 and Fig. S9†). As seen from the interaction analysis performed on all ACPs complexes, N5-Pan makes different hydrogen bonding patterns with the residues of the hydrophobic core of the ACP within the same group of simulated complexes, which implies that the sampling efficiency is significantly augmented by performing three independent simulations for the same complex. All simulated proteins equally favour the binding of the N5-Pan, since the calculated binding free energies are comparable to each other (Tables 2 and S1†). Still, this trend does not allow to conclude on the convergence of the calculations, but rather it corroborates the possibility of N5-Pan binding, and hence its inhibitory capacity.
Given that the hydrophobic core is occupied with N5-Pan, ACP does not show the sequestration of short-chain acyl moieties, which stay longer exposed to the solvent, and consequently more accessible to hydrolysis. For this type of inhibition, the following term is suggested: the stuffed pepper-like inhibition of the chain-flipping mechanism. A proposed name reflects the situation where there is no more room left to accommodate the acyl chain in the hydrophobic core of the ACP after N5-Pan is being bound (Fig. 5).
 | ||
| Fig. 5 A schematic representation showing the proposed stuffed pepper-like inhibition of the chain-flipping mechanism. The hexanoyl chain is not shown for clarity. | ||
Biological implications
ACP is certainly one of the most important protein–protein interaction hubs within the cell since is engaged in interactions with more than a dozen enzymes of the fatty acid synthase complex. A possible consequence of the N5-Pan binding in the hydrophobic core of the ACP could be the altered protein–protein interaction propensity towards the partner enzymes. This could be evident in the inability of the ACP to shuttle its cargo properly from one enzyme to the next one in the fatty acid biosynthesis pathway. However, the question of protein–protein interactions between various forms of ACP in complex with N5-Pan and partner enzymes of the FAS II should be addressed experimentally. Since the mammalian FAS I system possesses large macromolecular multidomain architecture36,37 while microbial FAS II is built up of separate proteins, any drug that would address separate enzymes of the bacterial type FAS I would not interfere with the protein domains of the mammalian FAS II. Considering architectural differences mentioned above, there has been a continuous effort in characterizing enzymes of microbial FAS, as well as novel compounds targeting them. Many compounds addressing the bacterial FAS enzymes have already been discovered.38 However, diversity in the enzymes and regulation of fatty acid synthesis in bacteria is enormous that we could point out a single organism that we could consider as a representative of bacteria as a whole or study as a model system for the fatty acid synthesis inhibition. Thus, the acyl carrier protein, central to fatty acid synthesis, which is omnipresent,39,40 and moreover, structurally and functionally conserved protein in bacteria, could be seen as a promising target through which we could control microbial viability. The hydrophobic pocket of microbial ACP could be a promising target for novel antibiotics, which structurally resembles the naturally sequestered intermediates of the fatty acid biosynthesis pathway. One should bear in mind that at the moment when E. coli cells are exposed to N5-Pan, E. coli proteome consists of various forms of acyl carrier proteins i.e. apo-CP, holo-CP and a variety of acyl-CP intermediates (3-oxoacyl-[acyl-carrier-protein], 3-hydroxyacyl-[acyl-carrier-protein], 2-trans-enoyl-[acyl-carrier-protein] and final saturated acyl-forms of ACP), since the fatty acid synthesis always operates in the viable cell. In this regard, N5-Pan could be seen as a potent inhibitor since it fits many protein targets that differ only slightly in the form of the prosthetic group and intermediate attached to it. Inhibitory potential of the N5-Pan could be realized through the additive effect since it addresses more than one target. Thus, any structural analog of 4′-phosphopantetheine prosthetic group with attached intermediate of the fatty acid synthesis pathway could be a potent inhibitor of the chain-flipping mechanism. In this way, the proper acyl chain sequestration as well as protein–protein communication, both essential in the fatty acid synthesis, could be defunct. However, after the discovery of N-alkylpantothenamides, structural modification of these compounds was directed towards the active site of PanK, an enzyme of the coenzyme A biosynthetic pathway, of E. coli,41S. aureus42,43 and P. falciparum44–46 in search of more potent inhibitors. The model of pantothenamide-induced inhibition of the bacterial growth proposed here questions this strategy of the N5-Pan structural modification directed exclusively towards the PanK active site and opens the possibility of a novel cellular target to be taken into account.The following experimental results published earlier corroborate the N5-Pan mode of action presented here. The pantothenate (a natural precursor for CoA biosynthesis) was added to the growth medium to test whether it would diminish the inhibitory effect of the N5-Pan on the bacterial growth.4 The addition of pantothenate to the growth medium did not diminish the inhibitory effect of the N5-Pan. Furthermore, overexpression of E. coli gene for PanK, pantothenate kinase, which catalyzes the first and regulatory step in CoA biosynthesis, failed to attenuate the inhibitory effect of the N5-Pan.4 These results indicate that N5-Pan does not inhibit the CoA biosynthetic pathway. Moreover, E. coli treated with the pantothenamides are incapable of incorporating [1-14C]-acetate to its membrane lipids, meaning that the fatty acid synthesis is inhibited.4 The supplementation of the growth medium with oleate failed to reverse growth inhibition by N5-Pan in E. coli strain ANS1. However, this was explained by the fact that the essential β-hydroxymyristic acid for lipid A synthesis in E. coli can not be provided from the growth medium.4 Therefore, a subsequent experiment selected the Gram-positive S. pneumoniae strain R6 against which N5-Pan was as effective as against E. coli strain. The addition of oleate to the growth medium increased the minimum inhibitory concentration (MIC) value by 4-fold, but was insufficient to restore full growth.4 Moreover, the addition of oleate and palmitate to the growth medium of S. aureus treated with pantothenamide, only allowed for the partial recovery of the bacterial growth.5 All these results and conclusions clearly indicate that the fatty acid synthesis is inhibited in the presence of the N5-Pan. Moreover, the growth curves indicated delayed toxicity of pantothenamides, showing a beginning of the slowdown in the growth of the bacterial culture during the third doubling period after the addition of the inhibitor. Interestingly, the authors themselves plainly pointed out:4 “The delayed toxicity of the pantothenamides suggested that the mechanism of the antibacterial activity required their incorporation into a cellular component rather than the direct inhibition of an essential enzyme for bacterial growth”.
Conclusions
In this study, widely employed in silico methods were used to test simple ideas, which have not been investigated earlier. Here is reported a new type of inhibition of microbial fatty acid synthesis that could serve as a completely new strategy for the design of novel antibiotics. It is demonstrated that acyl carrier protein can bind the prototypic N-pentylpantothenamide in the hydrophobic core, adopting the overall stuffed pepper look. As a result, the ACP with intercalated pantothenamide displays altered ability to sequester acyl chain in the hydrophobic core since it is occupied with N-pentylpantothenamide. Thus, acyl chain remains solvent-exposed on a longer time scale and susceptible to hydrolysis. Moreover, the ACP in complex with pantothenamide could change its chain-flipping mechanism for delivering acyl moiety to the downstream partner enzymes of the fatty acid synthesis pathway, which could lead to the decreased rates of fatty acid synthesis. Results presented here are in line with all previously published data and provide the missing link in the understanding of ambiguous experimental data,3–5 which to date have not explained how N5-Pan inhibits the growth of E. coli. From the published studies3–5 is known that N5-Pan is a substrate for PanK (a competitive inhibitor of PanK), and thus instead of CoA at the end of the CoA metabolic pathway, ethyldethia-CoA antimetabolite is being produced in the presence of N5-Pan.3 On the other hand, in E. coli, four genes, panB, panC, panD, and panE, encode four enzymes required for de novo pantothenic acid (physiological substrate for PanK) biosynthesis, which means that E. coli can overcome the inhibition of the CoA biosynthetic pathway eventually inhibited by the N5-Pan. Furthermore, the observed inhibition of fatty acid synthesis in the presence of N5-Pan was explained in the following way: ethyldethia-CoA metabolite was used as a substrate for the post-translational modification of ACP, which resulted in inactive ethyldethia-ACP incapable of delivering of a growing acyl chain in the fatty acid synthesis pathway.4 However, a subsequent study has shown that the ethyldethia-ACP is hydrolyzed in vivo,5 which means that E. coli can renew its apo-ACP pool which can be eventually used again for proper post-translational modification with a moiety derived from CoA. In addition to the rapid turnover, ACP is expressed constitutively in a cell. In other words, a physiologically active form of the ACP is always present in a cell. Thus, the inhibition of the bacterial growth is left unexplained. Therefore, the aim of this study was to investigate, bearing in mind the structural similarity between (a) the Ppant with attached acyl chain and (b) the N5-Pan, whether the N5-Pan could occupy the hydrophobic pocket of ACP, which naturally belongs to the Ppant prosthetic group with acyl chain attached to it.The proposed model of pantothenamide-induced inhibition of the bacterial growth raises many questions as to whether the structural modification of N-alkylpantothenamides should go in the direction of the optimal accommodation in the hydrophobic core of the ACP? Could we modify the N-pantothenamide structure in a way that we could obtain a tool for studying the chain-flipping mechanism as well? Moreover, how acyl carrier protein in complex with N5-Pan changes its interaction propensity towards the partner enzymes of the fatty acid biosynthesis pathway? Although the author has experience in working with similar proteins, and the previous results have already been experimentally confirmed and have been in line with experiments,47,48 unfortunately, computational methods alone are not sufficient and do not provide all answers to the questions that have been raised here. Therefore, the author is looking forward to passing the baton to experimental (bio)chemists in hopes of creating game-changing opportunities together.
Acknowledgements
A. M. would like to thank the University of Zagreb Computing Centre (SRCE) for generously granting computational resources on the ISABELLA cluster (http://isabella.srce.hr) and the CRO–NGI infrastructure (http://www.cro-ngi.hr). A. M. also wishes to thank the AMBER community for sharing the knowledge and providing the help (http://archive.ambermd.org/). Above all, A. M. wishes to thank Dr Biserka Kojić-Prodić, Prof. Dr Ita Gruić Sovulj and Dr Robert Vianello for their support.References
- G. Clifton, S. R. Bryant and C. G. Skinner, Arch. Biochem. Biophys., 1970, 137, 523–528 CrossRef CAS PubMed.
- C. Spry, C. Macuamule, Z. Lin, K. G. Virga, R. E. Lee, E. Strauss and K. J. Saliba, PLoS One, 2013, 8, e54974 CAS.
- E. Strauss and T. P. Begley, J. Biol. Chem., 2002, 277, 48205–48209 CrossRef CAS PubMed.
- Y. M. Zhang, M. W. Frank, K. G. Virga, R. E. Lee, C. O. Rock and S. Jackowski, J. Biol. Chem., 2004, 279, 50969–50975 CrossRef CAS PubMed.
- R. Leonardi, S. Chohnan, Y. M. Zhang, K. G. Virga, R. E. Lee, C. O. Rock and S. Jackowski, J. Biol. Chem., 2005, 280, 3314–3322 CrossRef CAS PubMed.
- J. Thomas and J. E. Cronan, Antimicrob. Agents Chemother., 2010, 54, 1374–1377 CrossRef CAS PubMed.
- E. Płoskoń, C. J. Arthur, A. L. Kanari, P. Wattana-amorn, C. Williams, J. Crosby, T. J. Simpson, C. L. Willis and M. P. Crump, Chem. Biol., 2010, 17, 776–785 CrossRef PubMed.
- D. I. Chan, T. Stockner, D. P. Tieleman and H. J. Vogel, J. Biol. Chem., 2008, 283, 33620–33629 CrossRef CAS PubMed.
- D. I. Chan, D. P. Tieleman and H. J. Vogel, Biochemistry, 2010, 49, 2860–2868 CrossRef PubMed.
- X. Qiu and C. A. Janson, Acta Cryst. D, 2004, 60, 1545–1554 Search PubMed.
- A. Roujeinikova, W. J. Simon, J. Gilroy, D. W. Rice, J. B. Rafferty and A. R. Slabas, J. Mol. Biol., 2007, 365, 135–145 CrossRef CAS PubMed.
- C. Nguyen, R. W. Haushalter, D. J. Lee, P. R. Markwick, J. Bruegger, G. Caldara-Festin, K. Finzel, D. R. Jackson, F. Ishikawa, B. O'Dowd, J. A. McCammon, S. J. Opella, S. C. Tsai and M. D. Burkart, Nature, 2014, 505, 427–431 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- P. Cieplak, W. D. Cornell, C. Bayly and P. A. Kollman, J. Comput. Chem., 1995, 16, 1357–1377 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- I. S. Joung and T. E. Cheatham, J. Phys. Chem. B, 2008, 112, 9020–9041 CrossRef CAS PubMed.
- H. J. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- R. J. Loncharich, B. R. Brooks and R. W. Pastor, Biopolymers, 1992, 32, 523–535 CrossRef CAS PubMed.
- A. W. Gotz, M. J. Williamson, D. Xu, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2012, 9, 1542–1555 CrossRef PubMed.
- S. Le Grand, A. W. Gotz and R. C. Walker, Comput. Phys. Commun., 2013, 184, 374–380 CrossRef CAS.
- R. Salomon-Ferrer, A. W. Goetz, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef CAS PubMed.
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Goetz, I. Kolossvai, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 2012, University of California, San Francisco, 2012 Search PubMed.
- J. P. Ryckaert, G. Ciccotti and H. J. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case and T. E. Cheatham III, Acc. Chem. Res., 2000, 33, 889–897 CrossRef CAS PubMed.
- D. Sitkoff, K. A. Sharp and B. Honig, J. Phys. Chem., 1994, 98, 1978–1988 CrossRef CAS.
- B. R. Miller III, T. D. McGee Jr, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef PubMed.
- D. A. Case, J. T. Berryman, R. M. Betz, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, T. Luchko, R. Luo, B. Madej, K. M. Merz, G. Monard, P. Needham, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, R. Salomon-Ferrer, C. L. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, D. M. York and P. A. Kollman, AMBER, University of California, San Francisco, 2015 Search PubMed.
- D. R. Roe and T. E. Cheatham III, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- M. Ester, H. P. Kriegel, J. Sander and X. Xu, A density-based algorithm for discovering clusters in large spatial databases with noise, in Proceedings of the Second International Conference on Knowledge Discovery and Data Mining (KDD-96), ed. E. Simoudis, J. Han and U. Fayyad, AAAI Press, Palo Alto, CA, 1996, pp. 226–231 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrodinger, LLC Search PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couc, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 13, 1605–1612 CrossRef PubMed.
- P. J. Turner, XMGRACE, Center for Coastal and Land-Margin Research Oregon Graduate Institute of Science and Technology, Beaverton, Oregon, USA, 2005 Search PubMed.
- T. Williams and C. Kelley, Gnuplot 4.5: an interactive plotting program, 2011 Search PubMed.
- T. Maier, M. Leibundgut and N. Ban, Science, 2008, 321, 1315–1322 CrossRef CAS PubMed.
- T. Maier, S. Jenni and N. Ban, Science, 2006, 311, 1258–1262 CrossRef CAS PubMed.
- J. B. Parsons and C. O. Rock, Curr. Opin. Microbiol., 2011, 14, 544–549 CrossRef CAS PubMed.
- P. Lu, C. Vogel, R. Wang, X. Yao and E. M. Marcotte, Nat. Biotechnol., 2007, 25, 117–124 CrossRef CAS PubMed.
- C. O. Rock and J. E. Cronan Jr, Methods Enzymol., 1981, 71, 341–351 CAS.
- K. G. Virga, Y. M. Zhang, R. Leonardi, R. A. Ivey, K. Hevener, H. W. Park, S. Jackowski, C. O. Rock and R. E. Lee, Bioorg. Med. Chem., 2006, 14, 1007–1020 CrossRef CAS PubMed.
- A. E. Choudhry, T. L. Mandichak, J. P. Broskey, R. W. Egolf, C. Kinsland, T. P. Begley, M. A. Seefeld, T. W. Ku, J. R. Brown, M. Zalacain and K. Ratnam, Antimicrob. Agents Chemother., 2003, 47, 2051–2055 CrossRef CAS PubMed.
- M. de Villiers, L. Barnard, L. Koekemoer, J. L. Snoep and E. Strauss, FEBS J., 2014, 281, 4731–4753 CrossRef CAS PubMed.
- C. Spry, C. L. L. Chai, K. Kirk and K. J. Saliba, Antimicrob. Agents Chemother., 2005, 49, 4649–4657 CrossRef CAS PubMed.
- M. de Villiers, C. Macuamule, C. Spry, Y. M. Hyun, E. Strauss and K. J. Saliba, ACS Med. Chem. Lett., 2013, 4, 784–789 CrossRef CAS PubMed.
- C. Spry, C. Macuamule, Z. Lin, K. G. Virga, R. E. Lee, E. Strauss and K. J. Saliba, PLoS One, 2013, 8, e54974 CAS.
- A. Maršavelski, S. Lesjak, M. Močibob, I. Weygand-Đurašević and S. Tomić, Mol. BioSyst., 2014, 10, 3207–3216 RSC.
- A. Maršavelski, M. Močibob, I. Gruić-Sovulj and R. Vianello, Phys. Chem. Chem. Phys., 2015, 17, 19030–19038 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra07430h |
| This journal is © The Royal Society of Chemistry 2016 |